
Simplistic combinatorial analyses are an honoured tradition in anti-evolutionary circles. Hoyle’s is the archetype of the combinatorial approach, and he gets a whole fallacy named after him for his trouble. The approach will be familiar – a string of length n composed of v different kinds of subunit is one point in a permutation space containing vn points in total. The chance of hitting any given sequence in one step, such as the one you have selected as ‘target’, is the reciprocal of that number. Exponentiation being the powerful tool it is, it takes only a little work with a calculator to assess the permutations available to the biological polymers DNA and protein and come up with some implausibly large numbers and conclude that Life – and, if you are feeling bold, evolution – is impossible.
Dryden, Thomson and White of Edinburgh University’s Chemistry department argue in this 2008 paper that not only is the combinatorial space of the canonical 20 L-acids much smaller than simplistically assumed, but more surprisingly, that it is sufficiently small to have been explored completely during the history of life on earth. The reason is that amino acids are not 20 completely different things, but a limited set of variants, each group of which varies mainly in shape and size, less so in chemical property. If one must use a textual analogy, this is not an alphabet of 20 different letters, but a half dozen or so, each of which can be rendered in several different fonts and pitches. But really, even roughly analogising them with language strings, and inferring restrictions on protein space from restrictions on locating or moving between viable strings in language space, fetches up against the rather obvious fact that amino acids are not letters, and proteins are not sentences or Shakespearean works.
There are obviously two parameters one can fiddle with to make the numbers look impressive – v and n.
- In the protein system in modern organisms, v=20 – there are 20 amino acids. Most proteins contain all 20. Where the asymmetry about the central (α) carbon atom permits the possibility of mirror image versions of the molecules, these occur invariably in their ‘left-handed’ version, but a ‘raw’ mixture generated non-biologically would give 39 different acids in all (one has no mirror image). There are even more possible acids, leading to a combinatorial explosion, in some imaginary ‘warm little pond’ with a mechanism gluing these things together at random
- As for n, the exponent in our space size parameter, it appears to be a feature of catalytic proteins (enzymes) that optimal structures are typically from several tens to thousands of subunits in length.
So – news to no-one – specific long proteins cannot be reached in a single step, especially if there are lots of different kinds of subunit to play with. A simple string of just 100 39-choice acids is one of 10159 permutations. If you think that’s still a bit cautious, stick the rest in. There are about 500 or so. 5001000? Too much even for Excel – the universe issues a #NUM!
Here are the flattened structures of the 20 amino acids:
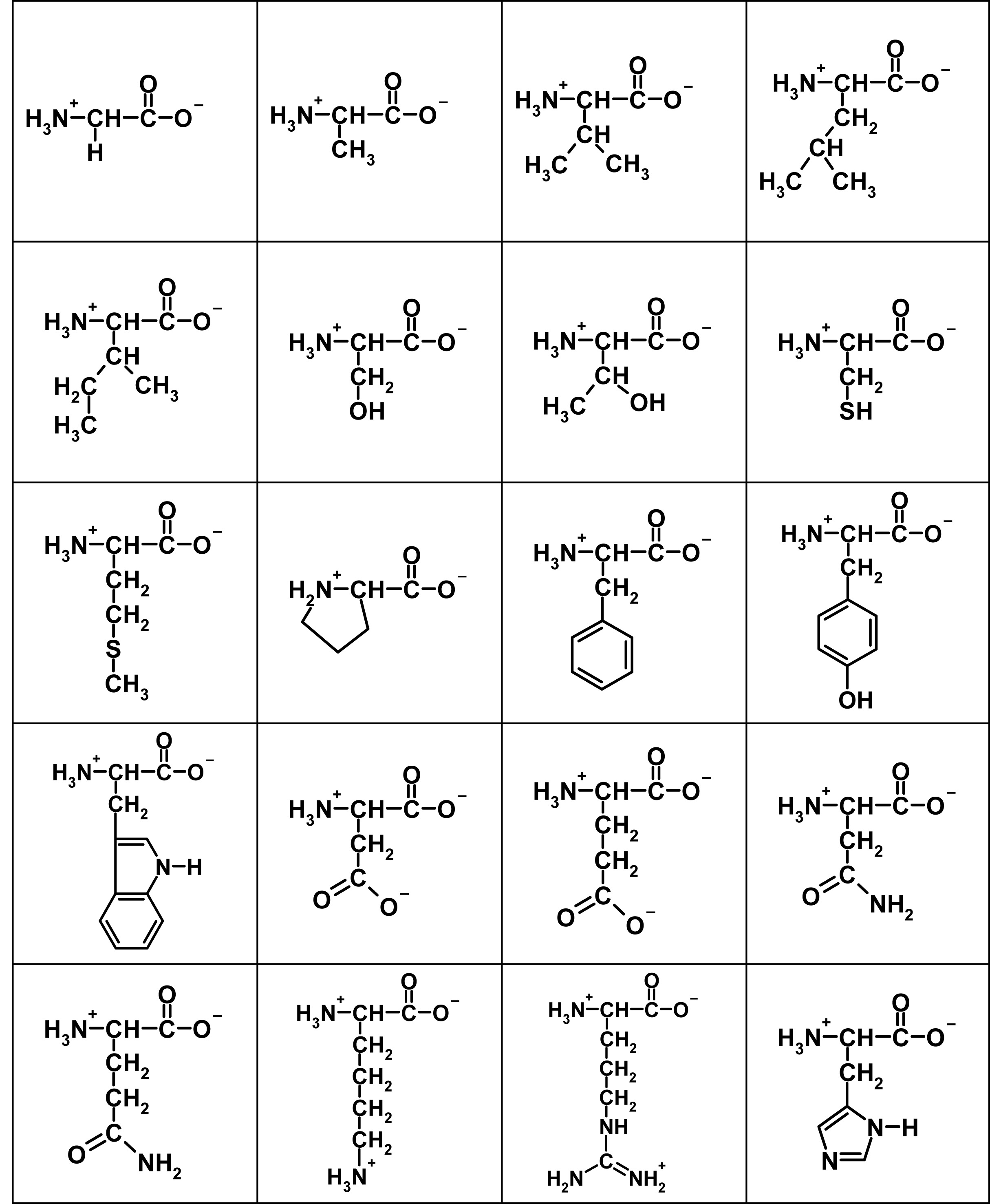
The salient features of these two-dimensional representations of 3D structure are the central (α) carbon with an H3N+– group to the left, a C-O– group to the right, and a side-chain depicted descending, with the simplest being a single hydrogen atom in Glycine top left. In a protein, these subunits are daisy-chained by linking the H3N+– group of one to the C-O– of another, eliminating two hydrogens and an oxygen (ie water) to form the peptide bond.
Because all acids are the L form – the D forms would have the side chain protruding upwards in the above chart – all side chains end up on the same side of the peptide-bond backbone. A freshly-synthesised peptide tends to shrivel up like a prude’s lips at gay Mardi Gras, as the various elements tussle for electrons with each other and with the surrounding water, and adopt the lowest-energy conformation. Like charges repel and unlike ones attract, while different side-groups have different affinity for the surrounding water and are variously repelled or attracted by it (hydrophobicity). The result is a complex fold adopted spontaneously and repeatably in the same physiological conditions. Where the peptide is an enzyme (not all peptides are), the fold brings the active site groups – usually just a handful of residues, often acting in tandem with a metal ion – into proximity, and the rest form a scaffold. The shape of this permits the entry of some substrates but excludes others, to give a degree of specificity, and its flexion can have a significant role in reaction kinetics.
Rather than name the acids, I will use a notation Row:Column to denote an acid by reference to the picture. Glycine, top left, is 1:1. Now, the substitutability of a given amino acid site depends very much on where it is, and what the substitution is. Substituting 1:3 with 1:4, for example, makes very little chemical difference. 1:4 is a little more hydrophobic (as this is not a strictly binary characteristic), and a little bigger (size, ditto!), so the minimum-energy conformation of the chain is slightly changed. But if the bulk of residues remain unchanged, the overall structure is much more constrained by that bulk than distorted by the substitution. Apart from a few key residues, such substitutions are unlikely to be catastrophic. But they do have the capacity to ‘tune’ the protein, in a beneficial or a detrimental direction, by the tiny steps postulated by the Darwinian mechanism. A poor fit can be progressively turned into a good fit, by blind ‘exploration’ of the protein neighbourhood. Other equivalent substitution groups can easily be identified from the chart – 2:2 for 2:3, 4:2 for 4:3, 5:2 for 5:3, even 1:2 for 2:1 etc. Substitution steps are not all equal in size.
Even though the sequential information can be rendered digitally, since it is modular, the mature folded peptide varies in an ‘analogue’ manner, by complex and continuous differentials between variants. There is no doubt that many substitutions are catastrophic, and many peptides cannot fold uniquely and repeatably, but this cannot be used to infer that all substitutions are doomed, nor that there is no way to gain a ‘toe-hold’ in the functional part of the space, which is essentially what Hoyle et al do. They assume, without evidence, that there is only one sequence in the whole of protein space that performs the function, and all the rest are duds. You can’t get there, and you can’t go anywhere else.
Any modern protein has been through the multi-generational filter of Natural Selection. It has likely explored its local protein space and this is the best it could come up with. While nearby variants may have been perfectly successful in their day, they cannot compete with the modern, ‘tuned’ version. So if they arise again, they are eliminated as detrimental mutations, where once they would have been top dog. This is a factor in the variation of both v and n. The smallest permutation space that includes a given modern protein includes all smaller spaces enfolded multiple times within it. Longer peptides appear to be closer to an optimum than shorter, but shorter strings drawn from a reduced amino acid set will still give peptides that perform biochemical functions. The range of functions, like the range of truly different acids, is limited. There are actually only about six. The enormous variety of the protein world comes from the wide variation of specificity of these fundamental reactions for different substrate molecules, controlled by the ‘wire frame’ shape and charge distribution. Even di- and tri-peptides (2 or 3 amino acids) can catalyse some of these basic reactions, though without the specificity of their giant cousins.
Nonetheless, protein space cannot be explored if it is not reasonably well-connected. One way to analyse its granularity with respect to function is to randomly sample a portion of it. It is necessary to restrict the search somewhat, because there are many more proteins that cannot fold reliably than can, but this is not the cheat it may appear. A 1993 paper by Kamtekar et al demonstrated a method of generating reliable α-helices (a very common motif in proteins) by simply dividing the amino acid set in a binary manner – polar and nonpolar – and creating a short, simple pattern based upon that found in natural protein helixes. The actual acid at any site was irrelevant, provided that it conformed to the appropriate polar/nonpolar nature of the natural residues. And these peptides folded. It is easy to see how a short stretch of such a fold – a dozen residues or so – can become a longer one, in a much bigger overall permutation space, simply by end-joining duplicates of the shorter sequences. And how such ‘modules’ can be moved around from protein to protein. All you actually need to gain a ‘toe-hold’ in the portion of the huge space occupied by working catalysts are one polar and one non-polar amino acid, in a pattern about a dozen residues long. 212=4096. Moving around that space is then a matter of various copy and paste mechanisms, not a fresh shake every time. Proteins are built from the substructures that work, not the many that don’t.
The extensive literature that cites the original Kamtekar paper, some shown on the same linked page, opens up a search space for the interested reader to explore the vast amount of work that has been done on the distribution of function in the world of randomly-generated, and randomly-swapped, peptide subsequences. The world of folded proteins is stuffed with function. This paper, for example, used a similar polar/nonpolar 14-acid patterning algorithm to generate a tiny portion of the space-of-all-peptides, which nonetheless contained functional analogues of 4 out of 27 natural peptides tried – a remarkable hit rate for essentially random sampling.
Hoyle’s imagined mechanism generates random, lengthy peptides from a raw amino acid mix. However, the probability of even a dipeptide in this ‘bumping into each other’ scenario is getting down towards zero. The ‘warm little pond’ is chemically naive; a strawman. Darwin (who coined the phrase) knew nothing of thermodynamics, nor protein. The free energy change associated with condensation/hydrolysis of the peptide bond means that it requires the input of energy to make it. The energy of motions of molecules in solution is not enough. Even with appropriate energy, having hit the jackpot once is insufficient. One has to retain that sequence, and this random process is not repeatable. So calculating ‘the probability of a protein’ by combinatorial means is irrelevant if that is not how it happened.
The problem does not go away, of course. We still have the thermodynamic and the repeat specification issues to contend with. A plausible solution to both is provided by the nucleic acids. It might appear that we have the same problem. Nucleic acid monomers need to polymerise, and for double helixes the specific ‘right-handed’ versions of the bases need to arise from a messier mix. This time, at least, the basic reaction is thermodynamically favoured. One of the nucleic acid monomers is ATP, the ‘energy currency’ of the cell. Although it is not a trivial matter to get the energy into ATP etc in the first place, once there, polymerisation can be driven by the available energy of the subunits. Besides their ‘energetic’ nature, an essential feature of the nucleic acid monomers is complementary pairing – A to T/U, and C to G. We could not realistically expect to start from a pure mix of right-handed monomers, neatly polymerising to form a single strand self-replicator to the exclusion of all contaminants. But this ability to complement, or hybridise, is a striking feature of nucleic acid strings. This isn’t just a mode of replication, but of stabilisation. Single strands will ‘fish’ their complement from a mixture and both strands are stabilised by the hydrogen bonding that ensues. Short chains made of consistently oriented subunits will complement more readily than those with mixed right- and left-handed monomers. In this way, non-replicating double helixes of complementary sequence would be the most stable form arising from a messier mix – a kind of purification process, ‘selecting’ those bases with optimal complementarity. Such structures cannot replicate, but this is a possible first step towards it without falling foul of the combinatorial issue – it is not necessary to locate a replicator sequence in search space before complementarity can evolve.
Like short peptides, short RNA and DNA strands have catalytic ability (ribozymes), and all the basic reactions are within their scope. One particularly relevant reaction is the ability to join an amino acid to a nucleic acid monomer, ATP, which can be accomplished by a ribozyme just 5 bases long. This is a central step in modern protein synthesis, the lone monomer now extended by an elaborate ‘tail’ arrangement – the tRNA molecule – and the joining now performed by a protein catalyst. Charging the acid in this way overcomes the thermodynamic barrier to peptide synthesis, because aminoacylated ATP has the energy to form a peptide bond where ‘bare’ amino acids do not. This gives an inkling of the mode by which Hoyle’s peptide space may have been actually accessed and explored. Short peptides formed by ribozymes from a limited acid library, with limited catalytic ability, may become longer and more specific and versatile by duplications and recombination of subunits. Meanwhile the acid library itself can extend by minor variations on the basic chemical themes until further variation is ultimately prevented by the extent of embedding of proteins in metabolism – it takes just one invariable site in an organism to freeze the underlying RNA codon assignment. There is a concomitant reduction of the catalytic role of RNA (though it remains, significantly, the catalyst for peptide bond formation). The one-handedness of the acid set would derive from the self-constraining one-handedness of nucleic acid monomers – because they are asymmetric, catalytic RNAs made from them are also asymmetric, and can only chain up one form of amino acid.
Excellent summary. I’ll have a go at reading the paper. Hope it answers the question in the title!
ETA the title in the paper “How much of protein sequence space has been explored by life on Earth?”
Yep – my title is slightly sarky!
I suppose it’s too much to expect kairosfocus to read the linked paper and offer comment?
Ho-hum.
I used to try to make the point at UD that functional proteins can be surprisingly insensitive to large variations in aa sequence; but got censored and banned for my pains.
Critical to the arguments made by Behe, Dembski, Myers, et al. is that a “blind” search is necessarily exhaustive. There simply can not be any natural means to limit the search space — hence the intelligent designer. Though they have never said as much, It’s as if they tacitly concede that “if” such natural mechanisms existed, then “Darwinism” would indeed be the better explanation.
As opposed to what TOE proposes which is that the search space is whatever can be reached by one mutation.
Maybe Joe’s TSZ syndication service will pick it up? 🙂
I’d be interested in WJM’s comments, since I was prompted to write the piece by a comment of his at UD. But it’s been bubbling away for years; you see that argument all the time.
The paper gives a nod to Axe, who has at least attempted to perform some experimental work. But there is a lot more work besides, much of it a short Google away. You can mess with proteins in all manner of ways and they still do something. Whether that something is physiologically useful is another matter, but the argument that there is only one way to skin a protein cat is illusory.
They are stuck somewhat between a rock and a hard place, because they (or many of them) agree that there is a place for the ‘Darwinian’ mechanism. The origin of variation, the tracking of small environmental fluctuations, the origin of certain groups of species by radiation. All of this must involve some variation (in the modern organism) of the protein repertoire. So space is locally explorable. The limitation is placed on any given genome and the variation available to it, but each step reveals a bit more of the landscape – a further set of possible steps.
Which is where islands of function come in. Eventually, genomes find certain areas of the space closed off to exploration; they bounce off a boundary. But there is no clear implementor of such a boundary, other than current occupation of adaptive peaks.
Tuned protein function certainly creates an ‘island’ – all steps are in the direction of detriment. But if it got there by Darwinian mechanisms, it is also Darwinian mechanisms that are drawing the boundary. It is not possible to vary the tuned function and create a new function from it, so if there were no mechanisms to sidestep that restriction ‘naturally’, a Designer might be the only option for change – take the sequence out of the organism, vary it in the lab, and stick it back somewhere else. But this is effectively gene duplication and mutation/recombination. There are natural mechanisms that do that. And if the adaptive landscape has changed, the previously ‘optimised’ state is no longer the best available. It’s no longer on an island anyway, and so needs no help getting off.
Exactly! The continually varying and multi-dimensional nature of the niche environment is something that some ID proponents find difficult to grasp
Also the fact that the evolving population is itself a changing aspect of the environment.
Thanks for this post btw! Very interesting paper. I might sticky it, to give people a chance to look at the paper.
Please. KF has more important things to be concerned with.
You dark triad people you.
Someone seems to be locked in the projection booth.
Ah yes (tappety-tap) the Dark Triad … lessee … (google google) … nope, doesn’t remind me of anyone at all! 😉
If KF has read the paper, or anything else on the matter, it is not clear from this.
A lengthy commission of Hoyle’s Fallacy, and attempt to analogise biological polymer space with communication strings, with no better justification than that both involve assemblies of length n consistng of v different subunits. Infinite monkeys have thus far failed to replicate more than a few words of Shakespeare, therefore … therefore absolutely nothing. I can draw 12 dots on a page and they don’t form a carbon nucleus either.
And this silly mistake:
If a beneficial variant arises, the ancestral state is the ‘inferior’ one. In SUBTRACTing these, NS leaves a population enriched in the beneficial variant – that’s addition. You can’t diminish one frequency without increasing another, since they must add up to 100%. SUBTRACTion and addition in populations are complementary, simultaneous processes.
Allan,
VJ Torley has responded to your post at UD:
Build me a protein – no guidance allowed! A response to Allan Miller and to Dryden, Thomson and White
I haven’t read it yet.
All that Torley just accomplished over at UD is another huge dump of the typical regurgitated ID/creationist misconceptions and “refutations.”
The molecules of life are not made of junkyard parts or letters of the alphabet.
It appears that the little high school level physics/chemistry calculation is still way over their heads; namely scale up the charge-to-mass ratios of protons and electrons to kilogram-size masses separated by one meter and then calculate the energy of interaction in joules and in megatons of TNT.
Then add the rules of quantum mechanics and, in the light of those answers, justify ID/creationist junkyard/alphabet calculations.
I’ve only skimmed. Given that many of the points were actually addressed, if indirectly, in the OP, it appears that Torley is not actually responding to my post at all, other than to abstract the fact that I made it and referenced Hoyle’s Fallacy and the ‘DTW’ paper. He simply restates the fallacy, and says it ‘s not a fallacy. It is.
I explain in some detail why it’s fallacious to equate protein construction with English sentences, but that analogy is beaten to death throughout the post and its quoted sources. He quotes four Creationist refutations of the paper which essentially cleave to the same fallacy (or set of fallacies): that one MUST regard all 20 amino acids as 20 completely different things, one MUST regard functional proteins as tiny pinpricks in the space of all possible proteins, one MUST regard the universality of the 20 acids in life as evidence that those 20 acids are the bare minimum for construction of functional proteins, one MUST regard only strings above a particular length threshold as ‘functional’, one MUST regard as functional only that part of protein space occupied by enzymes.
These are all unsound. It serves the ID position to beat them incessantly, and attempt to boost the numbers back up to their ‘impossible’ level, but there are no grounds for doing so.
I may construct a longer response, though I’d advise vjt not to hold his breath, as there is little of real substance I haven’t already addressed above. I’d be delighted if he would come here for a proper discussion, as I’m not that interested in booming across chasms under the asymmetric posting rules of the respective sites. I don’t read UD, so I won’t see his responses unless they are pointed out to me. If he has questions requiring answers, this is the place to ask them. I have, for reasons that were never made entirely clear, been banned at UD, so cannot respond there.
This, however, does pique my interest:
It is a fair question, and has, I believe, a fair answer. I addressed it to some degree in the OP; a more direct answer deserves the investment of time which I don’t, at the moment, have, but I will attempt to do so.
tjguy, in UD comments:
I addressed this point in the OP. Would it be too much to ask that UD-ers actually read the post that prompted Torley’s response, rather than just back-slapping him on his assumed takedown? I wonder what makes tjguy think that a mixture of left and right handed monomers would be catalytically inactive?
Clearly, a process has forced upon biology a settlement on homochiral acids, some time between the OoL (which may have had nothing to do with protein) and LUCA. The obvious candidate for such a process is the asymmetry of biological macromolecules themselves. However mixed their own subunit construction, the biological catalysts RNA and protein are asymmetric. They cannot build a pocket that can serve two different isomers and yet at the same time be specific. An enzyme – even one of mixed monomer construction – that works on L acids cannot work on D acids, and vice versa. As soon as such an asymmetry in monomer biosynthesis arises, it is reinforced. There is no point in catalysts specialising in the minority form. But the fundamental constraint on acid chirality is the shape of the ribosome.
Still haven’t gotten round to responding. But here is Axel, positively foaming at the mouth over what, to me, is simply an interesting (set of) puzzle(s).
What completely fails to be appreciated by Creationists the world over is the distinction between abiogenesis and evolution. The DTW paper is about evolution – traversal of protein space by replication – not landing in it to make replicators. Hoyle’s Fallacy contains two parts: the impossibility of an abiogenetic origin of proteins, and the impossibility of protein evolution. The first may be correct, but for reasons completely unconnected to Hoyle’s shaky combinatorial reasoning based upon modern protein sizes and components. It is not a given that the origin of life involved peptide bonds at all.
What quantum nonlocality has to do with anything, I’m not sure. Nor am I running from the possibility there may be a God. What a preposterous rationalisation of the ‘materialist’ mindset! Nobody possessed of any intellectual curiosity over there?
Rex Tugwell:
Having wondered if you might be missing something, you have evidently come to the conclusion: “Nah!”. Molecular biologists are just bullshitting you.
But one thing you may be missing is that the search of protein space is performed by changes to the underlying nucleic acid sequence. It isn’t performed by some other means and then converted into xNA, so successes don’t need to be ‘stored for later use’ – they’re already there. Further: it’s not apparent that the resident scientists are aware, but DNA and RNA are not completely different things. They are fully interconvertible. But for the presence or absence of a 2′ oxygen atom in the ribose and a methyl residue on one of the 4 bases, they are the same molecule. DNA just makes tighter helixes.
For those interested in a bit of biochemistry.
This is the straw that put all this discussion in motion. It starts with using a labeling fallacy against Hoyle.
Explain.
I must say, I didn’t start this line of reasoning! It is my answer to it. I believe the term “Hoyle’s fallacy” originated with Dawkins. The Wikipedia article originally had that title, at my time of writing (7 goddamned years ago, holds head in hands wondering what he’s done with his life!).
Call it something else if you like, it really doesn’t matter.
The space of functional nut and bolt pairs is practically infinite, but it doesn’t mean randomly generated 3D structures will result in a matching nut and bolt. Same for lock and keys, same for protein COMPLEXES — call it Quaternary structures if you like.
Look at all the domains the EZH2 protein alone needs! And then it’s part of the Polycomb Repression Complex to boot. Look at all the interface and contact points that have be positioned and oriented in 3D space between the proteins (like EZH2, SUZ12, EED) :
I think it would be a pretty daunting task to ask a protein engineering team to build proteins that would fold so nicely so as to fit so nicely together, much less act like a machine that can navigate to the right chromosome to the right stretch of DNA (the Hox cluster) to connect to the right histone (H3) and modified the right amino acid on the histone (amino acid 27). Boggles the mind. If our best engineers can’t do this putting their minds to it, why should we think random chance can do this? –and natural selection will be of little to no help, and may even prevent such evolution.
But nobody (other than creationists) claims that we are talking about randomly generated 3D structures.
It is better described as taking an already functioning structure, and tweaking it just a little. Then check whether that random tweak made it better or worse.
stcordova,
Not really addressing it either. Analogous systems are no guide to actual systems. I can stick nuts and bolts together any one of an infinity of ways and never get dehydrogenation of lactose.
stcordova,
Another answer – “If our best engineers can’t do this putting their minds to it” then it can’t be done by thinking about it!
stcordova,
Sal,
You know well chemistry is QM based…
Could proteins even fold, if we disrupted quantum correlations in the DNA?
stcordova,
It may be instructive to review VJ Torley’s response at UD, linked from my own response to his piece here. He basically did the same as you – find a picture of a modern protein, say it’s rilly rilly complex, show’s over.
If anything, I think a very large available search space should help, not hinder evolution. If proteins were limited to a short sequence length, they would have a much harder time at evolving complex structures, right? Imagine there was a limit of say 20 AA’s. A protein that long could only replace or remove AA’s in order to evolve, if it can also add AA’s the chances of finding a new functional sequence would be much higher, don’t you guys think?
Proteins fold in the complete absence of DNA, in a test tube, so yes.
dazz,
Yes, although a protein’s length is not related to function in a simple way. Some parts do catalysis, others act as a ‘wire frame’ to hold the catalytic part in place, others bind cofactors or membranes, or exclude unwanted substrates … looking at the whole and computing the hypothetical ‘search space’ for a protein that length is misleading, I think.
If you look at the protein in Sal’s (deliberately complex!) example above, you’ll see a lot of spirals. These are alpha helixes, one of the commonest motifs, and dead easy to form from all kinds of pretty random acid sequences. Acids have a ‘helix propensity’ – some are found in helixes more commonly than others, but all in that list can appear. Sometimes a less-ideal residue is compensated by those either side, so it’s contextual. If we took a naive view, we might think that a helix 20aa in length would have a 1 in 20^20 chance of forming. But, you can get a turn or two of helix from a huge number of different acid combinations. You can ‘tune’ a less-ideal turn. And you can end-join turns, creating longer stretches by duplication. The number 20^20 is pretty irrelevant – you don’t get there in one step, and there isn’t just one successful ‘hit’.
Btw, Happy New Year Allen. Thanks for the discussion.
From my Essential Cell Biology textbook by Alberts, it shows visually why CERTAIN proteins are improbable as a matter of principle. It’s desirable the electro static charges line up so the right part connects to the proper corresponding part. As Behe pointed out, it’s really bad if proteins randomly join to each other spontaneously. Functional complexes would then be broken by undesirable binding.
The PRC2 complex I showed is one example. Then there are homomeric proteins with Quaternary Structure like the homohexameric Helicase. Copies of itself have to fit together. Boggles the mind to think this can emerge randomly, and natural selection is outside the question since the the creature would be dead without it. If it’s dead, no selection. End of story.
The picture below is hexameric Helicase. Helicase is life critical if one considers cell replication life critical. Each of the colored segments is an identical copy of the helicase protein (I don’t know why they number the subunits 2-7 rather than 1-6, but oh well…). They connect together to make that quasi donut shape where DNA passes through the donut hole. I’ll provide a video in my next comment to show this incredibly well designed machine in action.
But the important point, just like the nuts and bolts problem, is the parts have to connect just right (with some degree of tolerance) to make a useful 3D structure that does something. Additionally, helicase needs an ATP domain to power it. So not only must it connect with other parts it needs an ATP domain in just the right spot to make the system work.
It requires some foresight as to how to put this complex together from 6 copies of the same protein!
I’m showing the space filling diagram for the helicase in this comment to show the snug fitting of the parts. I would presume, as Bruce Alberts suggests, the amino acids are positioned to make the parts desire to connect because of non-covalent binding on the interface surfaces and hydrophobic amino acids on the other surfaces to make connection possible. Oh, I didn’t say the helicase hexmaer needs to be assembled by other machines called “ring loaders” and “ring breakers”. So the helicase makes no sense without these, nor ATP to power it.
The next comment will show helicase in action. Boggles the mind to think a machine like this can assemble from random chemical soups. Forget about exploring protein space at all if life doesn’t even exist to explore it, and it won’t without something like helicase.
And please, let’s not appeal to phylogenetic methods as some sort of proof geometric problems shown here are easily solved by mutation and selection…
Here is a 2-minute video on Helicase. Boggles the mind how well conceived and designed this machine is:
https://youtu.be/bePPQpoVUpM
Heh. Wouldn’t be a day without Sal Cordova posting his usual argument from ignorance based personal incredulity.
Oh, and pictures. Got to have some pretty pictures. 🙂
stcordova,
Its also rather fortunate that in this universe, the space inside a doughnut hole actually exists.
Pity the poor universes with doughnut holes that don’t exist.
Life-its a piece of cake, er…doughnut!
stcordova,
Like I say – you’ve already pointed out how this can’t be done by thinking about it … 🤣 You would have it that this was done in a day, simultaneously in multitudes of functional individuals, many containing trillions of cells. I find that pretty boggling, but YBMV.
There’s nothing particularly boggling to me about initially weak interactions being tuned by evolution, such that the end state is a set of complementary contours. And clearly, you can get a hexamer by repeat duplication of a monomer, and ATP binding domains are ten-a-penny. You don’t start with the modern state, picked at random from protein space.
This works in the other dimension too. It is probable that the ancestral amino acid set was much smaller than 20. You only need about 3 different types of acid to make a catalytic globular protein, so a fairly small search space, length to the power three. As you add acids to the set, people would have it that evolution gets harder simply by virtue of the space getting bigger. And yet the richer set gives much more opportunity for subtlety and functional tuning, and opens up more pathways to bridge the space.
Allan Miller,
Thanks Allan, your first reply will take me a while to digest 😄, but yeah, that’s my intuition, and how I think it’s easy to see why it’s ridiculous to claim that a large search space is a problem for evolution. They just can’t stop thinking in terms of targets, oh well.
What? You have solved one of the life’s origins greatest mysteries? The amino acids, and the protein, formed spontaneously in the test tube without the need for information in DNA?
Finally! Congratulations!
The Thornton Lab has done extensive work showing how new gene regulatory modules can evolve (both the DNA binding spots for regulatory proteins, and the proteins themselves).
They have dusins of papers where they reconstruct the historical evolutionary trajectories that led to growth in the numbers of distinct regulatory proteins and their DNA binding targets, and how their regulatory mechanisms changed(how they changed in to respond to different hormones for example).
A particularly interesting and important, more recent one, is the one where they show that the gene-regulatory proteins that respond to different estrogen and steroid hormones, diverged from one ancestor by gene duplications. They even showed how the new function that evolved in this gene could have evolved along hundreds of different evolutionary trajectories without ever having to break the ancestral function, and without it being at all necessary for all the changes to occur simultaneously:
Starr TN, Picton LK, Thornton JW. Alternative evolutionary histories in the sequence space of an ancient protein. Nature. 2017 Sep 21;549(7672):409-413. DOI: 10.1038/nature23902
This also puts a lie to the oft repeated claim that biologists don’t test evolutionary inferences done using phylogenetics, with experiments. They do, the methods employed, called ancestor reconstruction, relies on phylogenetic methods, and provides a basis for being able to show that phylogenies aren’t just fantasies as Sal Cordova likes to claim. Phylogenetic methods have been tested empirically many times before(they’re also being used to show the evolution of language), and it’s been shown over and over again that they reproduce real genealogical histories to high accuracy. That means if you can show that your data exhibits a consistent phylogeny, you have very good reason to think it shows a real evolutionary and genealogical history. And that is what was done here, to show how a large family containing many distinct novel regulatory proteins that respond to estrogen and steroid hormones, evolved from a single ancestral gene. By gene duplication and divergence.
There is no mystery related to proteins being able to fold without DNA. DNA simply is not involved in making proteins fold, and nobody thinks it does.
Are you dyslexic? He says the proteins FOLD, as in they adopt their 3dimensonal structure, without help from DNA, he didn’t say whether the protein sequence is encoded in some DNA gene or not.
But also, yes, amino acids really can spontaneously polymerize into protein polymers under the right conditions, without having to be encoded in DNA. You didn’t know that? This is where you move the goalposts and start asking me to explain everything about the origin of life right? Where did X come from? How did Y occur? Huh? Huh? You don’t know so magic mustadunit.
Anyone who use arguments like Sal has been using against the prevailing view of evolution get accused of claiming without any further thought that it’s too complex therefore impossible. But equally it could be argued that defenders of the prevailing view haven’t thought deeply enough about the complexities and harmonious interactions involved. They focus in on processes that appear to them to involve simple structures being added together to make more complex structures in a similar way to lego construction. But building lego structures without any thought for the future would result in a static mass of lego structures with no further construction possible as there were only a finite amount of bricks.
Observed reality reveals that these living substances do not grow and function in this way. Dynamic processes of life and death must be balanced at all levels to maintain the viability of the whole. Protein complexes do not only require to be folded and combined into their so called quaternary structure (as Sal mentioned), a process which is fairly complex on its own without any further consideration. But these are not rigid structures. Proteins need to have the suitable strength, intrinsic mobility, extrinsic mobility and the lifespan necessary to carry out their various functions. All this must work in a coordinated way with all the other processes going on in the cell in order to ensure the viability of the cell though its natural lifespan. And as in proteins the same goes for cells, and for organs and organisms, and ecosystems and life as a whole. Earthly life can only survive and evolve because it maintains a dynamic balance at and between all levels. Any runaway destabilisation of harmony at a lower level extinguishes the higher level and vice versa.
I’m sure Sal has put a great deal of thought into the dynamic, interweaving complexities of life. Without the restraints and checks of forward planning positive feedback will bring a halt to any complex system.
Me? Really?
Here it is… You don’t know but magic mustdunit… All the magic imaginable, but one…
It’s quite educational to observe people to try to find the answer because they don’t want to hear the obvious one…
You can explode with anger now…
Getting function from randomness is a piece of cake…all one needs is magic… or a miracle… as long as it is not from one source…
Isn’t it obvious? What a relief! 😉 No accountibility…
No, that’s false. What they get rightly accused of is to merely assert that conclusion. They never actually show that conclusion to be true.
Just look at how Sal actually argues, there are no demonstrations or arguments to the truth of his assertions. He just lists a bunch of claims and then declares his incredulity, without his fact-claims being shown to be true, nor that his incredulity is justified.
Look at how he argues his case. First he says:
“The space of functional nut and bolt pairs is practically infinite, but it doesn’t mean randomly generated 3D structures will result in a matching nut and bolt.”
It’s formulated in a weird way. Does the size of the total space autmatically mean “randomly generated 3D structures will result in a match”?
No, that is not guaranteed. But it is also not guaranteed, nor even implied, that no matches will be in that space, nor does that by itself imply anything about how likely such matches are.
Then he goes on to state this:
Look at all the domains the EZH2 protein alone needs! And then it’s part of the Polycomb Repression Complex to boot. Look at all the interface and contact points that have be positioned and oriented in 3D space between the proteins (like EZH2, SUZ12, EED) :”
Okay, I’m looking at it. Yeah, and? What am I supposed to see? This is where Sal’s chain unhinges, because we jump straight into Sal’s incredulity. He doesn’t SHOW his incredulity to be justified, he doesn’t SHOW that X evolving is impossible or even unlikely. He just gives that list of facts, and then declares
“I think it would be a pretty daunting task to ask a protein engineering team to build proteins that would fold so nicely so as to fit so nicely together, much less act like a machine that can navigate to the right chromosome to the right stretch of DNA (the Hox cluster) to connect to the right histone (H3) and modified the right amino acid on the histone (amino acid 27).”
Oh, Sal thinks it would be a “daunting task” to ask a protein engineering team to do? So what? What follows from that? Nothing. It’s just a statement about what Sal thinks is a daunting task to a protein engineering team.
He gives no reason for why these systems can’t evolve. There’s no argument here.
“Boggles the mind.”
His mind is now boggled. Why didn’t he tell us this to begin with? Evolution MUST be false then.
“If our best engineers can’t do this putting their minds to it why should we think random chance can do this?”
Well first of all it isn’t established our best engineers can’t do this. It’s not even established a mediocre engineer can’t do it. But let’s just pretend that’s true, what follows? Nothing. That tells us nothing about whether it can evolve.
I can’t lift a tectonic plate, but the pressures inside the Earth can, and built a mountain range. Whatever human beings can personally accomplish is flatly irrelevant to what can occur in nature. Evolution isn’t employing human engineers to make proteins. Proteins evolve by mutations and natural selection.
” –and natural selection will be of little to no help, and may even prevent such evolution.”
And this is where Sal Cordova just DECLARES that evolution will be of little to no help, and may even prevent evolution. He just SAYS this. He gives NO argument or supporting evidence. None whatsoever.
His whole post was vacuous nonsense. And you’re impressed by the “deep thought” you perceive is put into this shit?
That’s cute. I’m pretty sure that he has not, if the above empty rhetoric is any indication.
So you declare, with zero supporting evidence or reason. Nor is it at all clear what this even has to do with biological evolution.
You both speak in vague generalities and then declare some grandiose conclusion from it without ever showing that it is true or even implied.
Oh look at how complex it is, look at all the things that have to happen, look at all the things that interact. Oh how, oh how, oh how, how could it possibly evolve?
Well if you read the paper I linked above, they will tell not only how it could evolve, but give a concrete real-world example based on laboratory experiments, where they figured out how it DID evolve. And go even further to show how it could ALSO have evolved by entirely different ways than occurred in history.
You have nothing but empty rhetoric, and meanwhile real evolutionary biologists doing actual work in the field and in the lab are showing with examples how evolution happens. All you can do is to stand on the sidelines and scream, stamp your feet, and insist you can’t possibly accept or believe it. It’s pretty clear that, at this stage, after so much time having had these things explained to you over and over again for decades, you simply don’t want to.
Neil Rickert,
Biological control functions work or they don’t. You can either recycle a protein or you cannot. All the chickens and eggs need to be there at once.
Evidence please that magic or miracles are required! Got any? No? Oh well.
To further clarify, in this post to Sal, I was referring to the conversation Sal and I have had (and Gordon Davidson) on many different OPs regarding quantum information, Q correlations etc. in DNA.
There is now solid proof, that quantum mechanics (quantum jitters) is involved in mutations suggesting a possibility of directed mutations, such as adaptive mutation…etc.
The ongoing conversations relates to quantum information in DNA; if DNA contains quantum information, the disruption of that information could lead to interruptions of DNA transcription; i.e. translation errors . This can have a direct effect the amino-acid sequence, the protein fold, protein function, protein misfolding etc.
This issue is separate from the one I joked about with Allan Miller
but in the board sense of evolution of proteins and the origins of life, it still applies…
People with set world view will never learn anything because they simply do not want to… It’s fine but it’s hard to respect that…