
Simplistic combinatorial analyses are an honoured tradition in anti-evolutionary circles. Hoyle’s is the archetype of the combinatorial approach, and he gets a whole fallacy named after him for his trouble. The approach will be familiar – a string of length n composed of v different kinds of subunit is one point in a permutation space containing vn points in total. The chance of hitting any given sequence in one step, such as the one you have selected as ‘target’, is the reciprocal of that number. Exponentiation being the powerful tool it is, it takes only a little work with a calculator to assess the permutations available to the biological polymers DNA and protein and come up with some implausibly large numbers and conclude that Life – and, if you are feeling bold, evolution – is impossible.
Dryden, Thomson and White of Edinburgh University’s Chemistry department argue in this 2008 paper that not only is the combinatorial space of the canonical 20 L-acids much smaller than simplistically assumed, but more surprisingly, that it is sufficiently small to have been explored completely during the history of life on earth. The reason is that amino acids are not 20 completely different things, but a limited set of variants, each group of which varies mainly in shape and size, less so in chemical property. If one must use a textual analogy, this is not an alphabet of 20 different letters, but a half dozen or so, each of which can be rendered in several different fonts and pitches. But really, even roughly analogising them with language strings, and inferring restrictions on protein space from restrictions on locating or moving between viable strings in language space, fetches up against the rather obvious fact that amino acids are not letters, and proteins are not sentences or Shakespearean works.
There are obviously two parameters one can fiddle with to make the numbers look impressive – v and n.
- In the protein system in modern organisms, v=20 – there are 20 amino acids. Most proteins contain all 20. Where the asymmetry about the central (α) carbon atom permits the possibility of mirror image versions of the molecules, these occur invariably in their ‘left-handed’ version, but a ‘raw’ mixture generated non-biologically would give 39 different acids in all (one has no mirror image). There are even more possible acids, leading to a combinatorial explosion, in some imaginary ‘warm little pond’ with a mechanism gluing these things together at random
- As for n, the exponent in our space size parameter, it appears to be a feature of catalytic proteins (enzymes) that optimal structures are typically from several tens to thousands of subunits in length.
So – news to no-one – specific long proteins cannot be reached in a single step, especially if there are lots of different kinds of subunit to play with. A simple string of just 100 39-choice acids is one of 10159 permutations. If you think that’s still a bit cautious, stick the rest in. There are about 500 or so. 5001000? Too much even for Excel – the universe issues a #NUM!
Here are the flattened structures of the 20 amino acids:
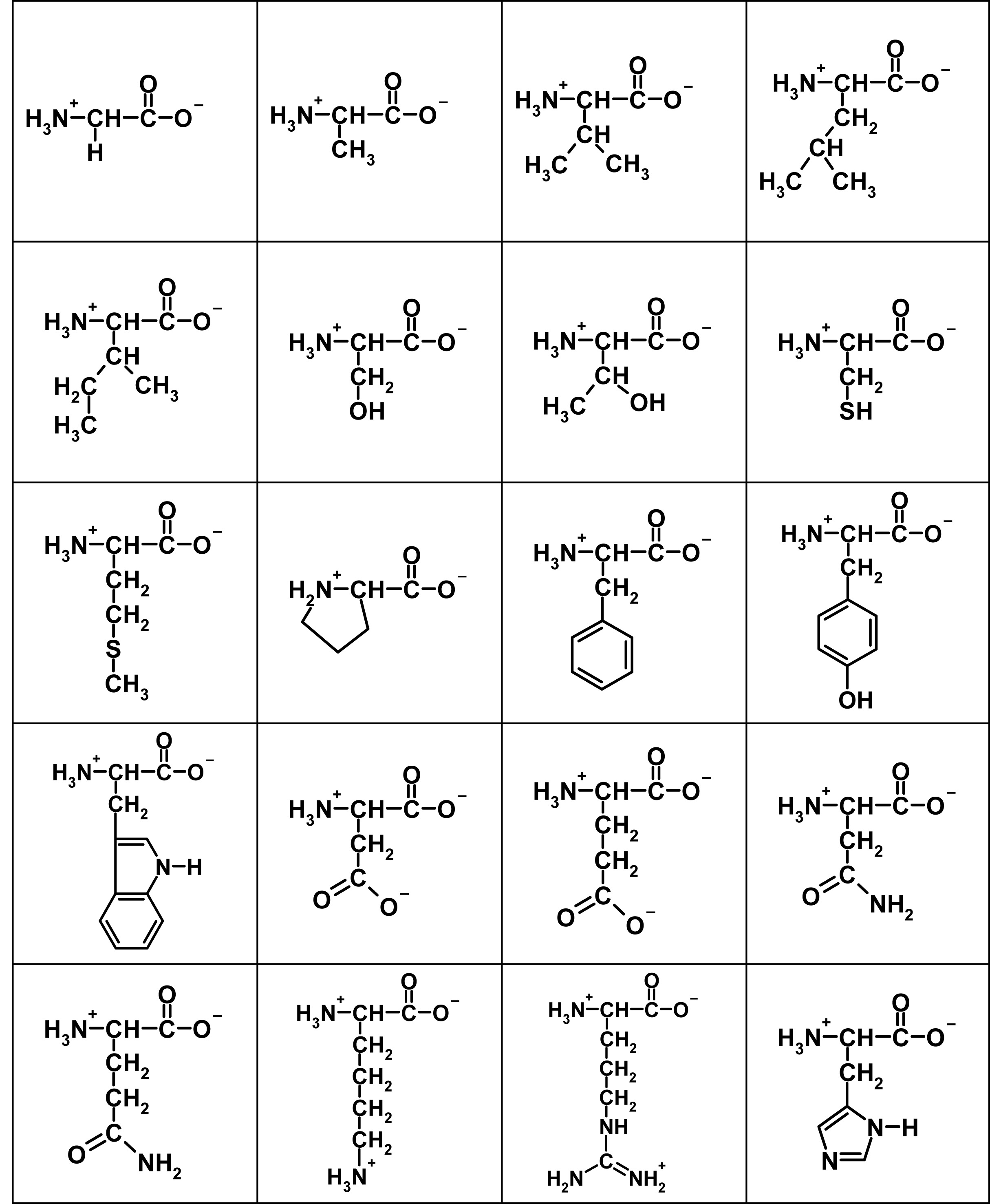
The salient features of these two-dimensional representations of 3D structure are the central (α) carbon with an H3N+– group to the left, a C-O– group to the right, and a side-chain depicted descending, with the simplest being a single hydrogen atom in Glycine top left. In a protein, these subunits are daisy-chained by linking the H3N+– group of one to the C-O– of another, eliminating two hydrogens and an oxygen (ie water) to form the peptide bond.
Because all acids are the L form – the D forms would have the side chain protruding upwards in the above chart – all side chains end up on the same side of the peptide-bond backbone. A freshly-synthesised peptide tends to shrivel up like a prude’s lips at gay Mardi Gras, as the various elements tussle for electrons with each other and with the surrounding water, and adopt the lowest-energy conformation. Like charges repel and unlike ones attract, while different side-groups have different affinity for the surrounding water and are variously repelled or attracted by it (hydrophobicity). The result is a complex fold adopted spontaneously and repeatably in the same physiological conditions. Where the peptide is an enzyme (not all peptides are), the fold brings the active site groups – usually just a handful of residues, often acting in tandem with a metal ion – into proximity, and the rest form a scaffold. The shape of this permits the entry of some substrates but excludes others, to give a degree of specificity, and its flexion can have a significant role in reaction kinetics.
Rather than name the acids, I will use a notation Row:Column to denote an acid by reference to the picture. Glycine, top left, is 1:1. Now, the substitutability of a given amino acid site depends very much on where it is, and what the substitution is. Substituting 1:3 with 1:4, for example, makes very little chemical difference. 1:4 is a little more hydrophobic (as this is not a strictly binary characteristic), and a little bigger (size, ditto!), so the minimum-energy conformation of the chain is slightly changed. But if the bulk of residues remain unchanged, the overall structure is much more constrained by that bulk than distorted by the substitution. Apart from a few key residues, such substitutions are unlikely to be catastrophic. But they do have the capacity to ‘tune’ the protein, in a beneficial or a detrimental direction, by the tiny steps postulated by the Darwinian mechanism. A poor fit can be progressively turned into a good fit, by blind ‘exploration’ of the protein neighbourhood. Other equivalent substitution groups can easily be identified from the chart – 2:2 for 2:3, 4:2 for 4:3, 5:2 for 5:3, even 1:2 for 2:1 etc. Substitution steps are not all equal in size.
Even though the sequential information can be rendered digitally, since it is modular, the mature folded peptide varies in an ‘analogue’ manner, by complex and continuous differentials between variants. There is no doubt that many substitutions are catastrophic, and many peptides cannot fold uniquely and repeatably, but this cannot be used to infer that all substitutions are doomed, nor that there is no way to gain a ‘toe-hold’ in the functional part of the space, which is essentially what Hoyle et al do. They assume, without evidence, that there is only one sequence in the whole of protein space that performs the function, and all the rest are duds. You can’t get there, and you can’t go anywhere else.
Any modern protein has been through the multi-generational filter of Natural Selection. It has likely explored its local protein space and this is the best it could come up with. While nearby variants may have been perfectly successful in their day, they cannot compete with the modern, ‘tuned’ version. So if they arise again, they are eliminated as detrimental mutations, where once they would have been top dog. This is a factor in the variation of both v and n. The smallest permutation space that includes a given modern protein includes all smaller spaces enfolded multiple times within it. Longer peptides appear to be closer to an optimum than shorter, but shorter strings drawn from a reduced amino acid set will still give peptides that perform biochemical functions. The range of functions, like the range of truly different acids, is limited. There are actually only about six. The enormous variety of the protein world comes from the wide variation of specificity of these fundamental reactions for different substrate molecules, controlled by the ‘wire frame’ shape and charge distribution. Even di- and tri-peptides (2 or 3 amino acids) can catalyse some of these basic reactions, though without the specificity of their giant cousins.
Nonetheless, protein space cannot be explored if it is not reasonably well-connected. One way to analyse its granularity with respect to function is to randomly sample a portion of it. It is necessary to restrict the search somewhat, because there are many more proteins that cannot fold reliably than can, but this is not the cheat it may appear. A 1993 paper by Kamtekar et al demonstrated a method of generating reliable α-helices (a very common motif in proteins) by simply dividing the amino acid set in a binary manner – polar and nonpolar – and creating a short, simple pattern based upon that found in natural protein helixes. The actual acid at any site was irrelevant, provided that it conformed to the appropriate polar/nonpolar nature of the natural residues. And these peptides folded. It is easy to see how a short stretch of such a fold – a dozen residues or so – can become a longer one, in a much bigger overall permutation space, simply by end-joining duplicates of the shorter sequences. And how such ‘modules’ can be moved around from protein to protein. All you actually need to gain a ‘toe-hold’ in the portion of the huge space occupied by working catalysts are one polar and one non-polar amino acid, in a pattern about a dozen residues long. 212=4096. Moving around that space is then a matter of various copy and paste mechanisms, not a fresh shake every time. Proteins are built from the substructures that work, not the many that don’t.
The extensive literature that cites the original Kamtekar paper, some shown on the same linked page, opens up a search space for the interested reader to explore the vast amount of work that has been done on the distribution of function in the world of randomly-generated, and randomly-swapped, peptide subsequences. The world of folded proteins is stuffed with function. This paper, for example, used a similar polar/nonpolar 14-acid patterning algorithm to generate a tiny portion of the space-of-all-peptides, which nonetheless contained functional analogues of 4 out of 27 natural peptides tried – a remarkable hit rate for essentially random sampling.
Hoyle’s imagined mechanism generates random, lengthy peptides from a raw amino acid mix. However, the probability of even a dipeptide in this ‘bumping into each other’ scenario is getting down towards zero. The ‘warm little pond’ is chemically naive; a strawman. Darwin (who coined the phrase) knew nothing of thermodynamics, nor protein. The free energy change associated with condensation/hydrolysis of the peptide bond means that it requires the input of energy to make it. The energy of motions of molecules in solution is not enough. Even with appropriate energy, having hit the jackpot once is insufficient. One has to retain that sequence, and this random process is not repeatable. So calculating ‘the probability of a protein’ by combinatorial means is irrelevant if that is not how it happened.
The problem does not go away, of course. We still have the thermodynamic and the repeat specification issues to contend with. A plausible solution to both is provided by the nucleic acids. It might appear that we have the same problem. Nucleic acid monomers need to polymerise, and for double helixes the specific ‘right-handed’ versions of the bases need to arise from a messier mix. This time, at least, the basic reaction is thermodynamically favoured. One of the nucleic acid monomers is ATP, the ‘energy currency’ of the cell. Although it is not a trivial matter to get the energy into ATP etc in the first place, once there, polymerisation can be driven by the available energy of the subunits. Besides their ‘energetic’ nature, an essential feature of the nucleic acid monomers is complementary pairing – A to T/U, and C to G. We could not realistically expect to start from a pure mix of right-handed monomers, neatly polymerising to form a single strand self-replicator to the exclusion of all contaminants. But this ability to complement, or hybridise, is a striking feature of nucleic acid strings. This isn’t just a mode of replication, but of stabilisation. Single strands will ‘fish’ their complement from a mixture and both strands are stabilised by the hydrogen bonding that ensues. Short chains made of consistently oriented subunits will complement more readily than those with mixed right- and left-handed monomers. In this way, non-replicating double helixes of complementary sequence would be the most stable form arising from a messier mix – a kind of purification process, ‘selecting’ those bases with optimal complementarity. Such structures cannot replicate, but this is a possible first step towards it without falling foul of the combinatorial issue – it is not necessary to locate a replicator sequence in search space before complementarity can evolve.
Like short peptides, short RNA and DNA strands have catalytic ability (ribozymes), and all the basic reactions are within their scope. One particularly relevant reaction is the ability to join an amino acid to a nucleic acid monomer, ATP, which can be accomplished by a ribozyme just 5 bases long. This is a central step in modern protein synthesis, the lone monomer now extended by an elaborate ‘tail’ arrangement – the tRNA molecule – and the joining now performed by a protein catalyst. Charging the acid in this way overcomes the thermodynamic barrier to peptide synthesis, because aminoacylated ATP has the energy to form a peptide bond where ‘bare’ amino acids do not. This gives an inkling of the mode by which Hoyle’s peptide space may have been actually accessed and explored. Short peptides formed by ribozymes from a limited acid library, with limited catalytic ability, may become longer and more specific and versatile by duplications and recombination of subunits. Meanwhile the acid library itself can extend by minor variations on the basic chemical themes until further variation is ultimately prevented by the extent of embedding of proteins in metabolism – it takes just one invariable site in an organism to freeze the underlying RNA codon assignment. There is a concomitant reduction of the catalytic role of RNA (though it remains, significantly, the catalyst for peptide bond formation). The one-handedness of the acid set would derive from the self-constraining one-handedness of nucleic acid monomers – because they are asymmetric, catalytic RNAs made from them are also asymmetric, and can only chain up one form of amino acid.
OK, It seems that nobody would like to answer this question, so I’m going to ask it again:
1. Is the speed light constant, or not?
2. If the speed of light were not constant, as I have suggested it here more than once, what would be the implications?
I’m sure all those skeptics here have the experimental evidence and the mathematical proof to support their views, rather than begging for fair-mindedness… or resorting to cheep shots of all sorts, like holocaust denier, antisemitism etc…
This is really funny, but I’m not a geocentrist, though newton has tried to convince me that the Bible literary teaches it…
All I know is that relativity doesn’t prohibit geocentrism and Einstein has mentioned it on many occasions. (More on this soon) 😉
What I really enjoy is the atheists/materialists and phony theists have convinced themselves that geocentrism shouldn’t be the preferred view , or nobody should be thinking it, because it places the Earth in the special location in the solar system, or even in the center of the universe and that makes them look bad, because of their preconceived world view… 😉
Where does the absurdity end?
Maybe it’s not that nobody wants to answer the question. Maybe most people don’t know.
I think it isn’t. As far as I remember, the speed of light changes a tiny bit when confronted with different media. This is why light refracts when entering water, or a prism, at an angle. Some wavelengths are slowed down a bit more than others.
It would refract?
This paragraph doesn’t make sense. I don’t know if I should try and ask what it means, or just ignore it. For example, why would this be either have the proofs or beg (beg?) for fair mindedness? Cheep shots? What would Holocaust denialism and antisemitism have to do with whether the speed of light is constant?
Entropy,
J-Mac is on the wrong thread, is all.
If you both consent, I can try moving these comments to the “No Copernican Principle…” thread.
.. he declared with great conviction. Being a God himself, he needed only speak and it was so.
That’s all right by me. This thread is derailed either way though.
We have evidence of teleology in nature by observing the actions of animals. And of course of conscious teleology through our own actions, and we are part of nature.
I don’t see the creation of form in either development or evolution in mechanistic terms. Although it obviously has mechanical aspects the creativity does not spring from the mechanics IMO.
It is your assumption that mutations drive evolution, not mine.
Up to you…
Sorry, I just realized both of my comments may belong on CP thread…
You can move both…
I’ll assume we are talking about the speed of light in a vacuum, because it varies in other media. And, note there there isn’t any true vacuum, so this is an idealization.
And then we need to know whether you are asking about the speed of light according to Newtonian standards, or according to relativistic standards.
With respect to Newtonian standards, the Michelson-Morley experiment pretty much showed that any variation will be small enough that it is hard to measure.
With respect to relativistic standards, the speed of light (in vacuum) is constant. That constancy is built into relativistic standards. And relativistic standards make better predictions, which is why physics uses them.
The most likely explanation is that you flubbed the measurement. It is also possible that there is something amiss in physics, but that’s far less likely.
Hmmm…
But what are the relativistic standards for two photons moving in the opposite direction?
BTW: You should probably move this comment to the CP OP
It’s not an assumption, it is a demonstrable fact that one of the reasons evolution occurs is that mutations happen.
I know what you’re going to say, you’re going to feed me some nonsense about the organism being “considered holistically” somehow driving it’s own evolution, and so, you will attempt to claim, it is the organism itself causing the mutations.
Here’s the problem: It’s demonstrably bullshit.
The replication machinery can be removed from the organism, and tested in vitro, and in total isolation of the rest of the the cell, the DNA polymerase enzyme will make mutations randomly as it replicates the DNA.
Heck, the cell even has numerous enzymes that attempt to correct DNA mismatches, double-strand breaks, spontaneous deaminations and so on.
But these systems too have intrinsic rates of error, so mutations still slip through. And all of them, without exception, have been demonstrated to function and make occasional, random errors, in the total absence of the rest of the cells.
So no, the cell isn’t some “holistic” thing that drives it’s own evolution. That’s wrong, and nonsense.