
Simplistic combinatorial analyses are an honoured tradition in anti-evolutionary circles. Hoyle’s is the archetype of the combinatorial approach, and he gets a whole fallacy named after him for his trouble. The approach will be familiar – a string of length n composed of v different kinds of subunit is one point in a permutation space containing vn points in total. The chance of hitting any given sequence in one step, such as the one you have selected as ‘target’, is the reciprocal of that number. Exponentiation being the powerful tool it is, it takes only a little work with a calculator to assess the permutations available to the biological polymers DNA and protein and come up with some implausibly large numbers and conclude that Life – and, if you are feeling bold, evolution – is impossible.
Dryden, Thomson and White of Edinburgh University’s Chemistry department argue in this 2008 paper that not only is the combinatorial space of the canonical 20 L-acids much smaller than simplistically assumed, but more surprisingly, that it is sufficiently small to have been explored completely during the history of life on earth. The reason is that amino acids are not 20 completely different things, but a limited set of variants, each group of which varies mainly in shape and size, less so in chemical property. If one must use a textual analogy, this is not an alphabet of 20 different letters, but a half dozen or so, each of which can be rendered in several different fonts and pitches. But really, even roughly analogising them with language strings, and inferring restrictions on protein space from restrictions on locating or moving between viable strings in language space, fetches up against the rather obvious fact that amino acids are not letters, and proteins are not sentences or Shakespearean works.
There are obviously two parameters one can fiddle with to make the numbers look impressive – v and n.
- In the protein system in modern organisms, v=20 – there are 20 amino acids. Most proteins contain all 20. Where the asymmetry about the central (α) carbon atom permits the possibility of mirror image versions of the molecules, these occur invariably in their ‘left-handed’ version, but a ‘raw’ mixture generated non-biologically would give 39 different acids in all (one has no mirror image). There are even more possible acids, leading to a combinatorial explosion, in some imaginary ‘warm little pond’ with a mechanism gluing these things together at random
- As for n, the exponent in our space size parameter, it appears to be a feature of catalytic proteins (enzymes) that optimal structures are typically from several tens to thousands of subunits in length.
So – news to no-one – specific long proteins cannot be reached in a single step, especially if there are lots of different kinds of subunit to play with. A simple string of just 100 39-choice acids is one of 10159 permutations. If you think that’s still a bit cautious, stick the rest in. There are about 500 or so. 5001000? Too much even for Excel – the universe issues a #NUM!
Here are the flattened structures of the 20 amino acids:
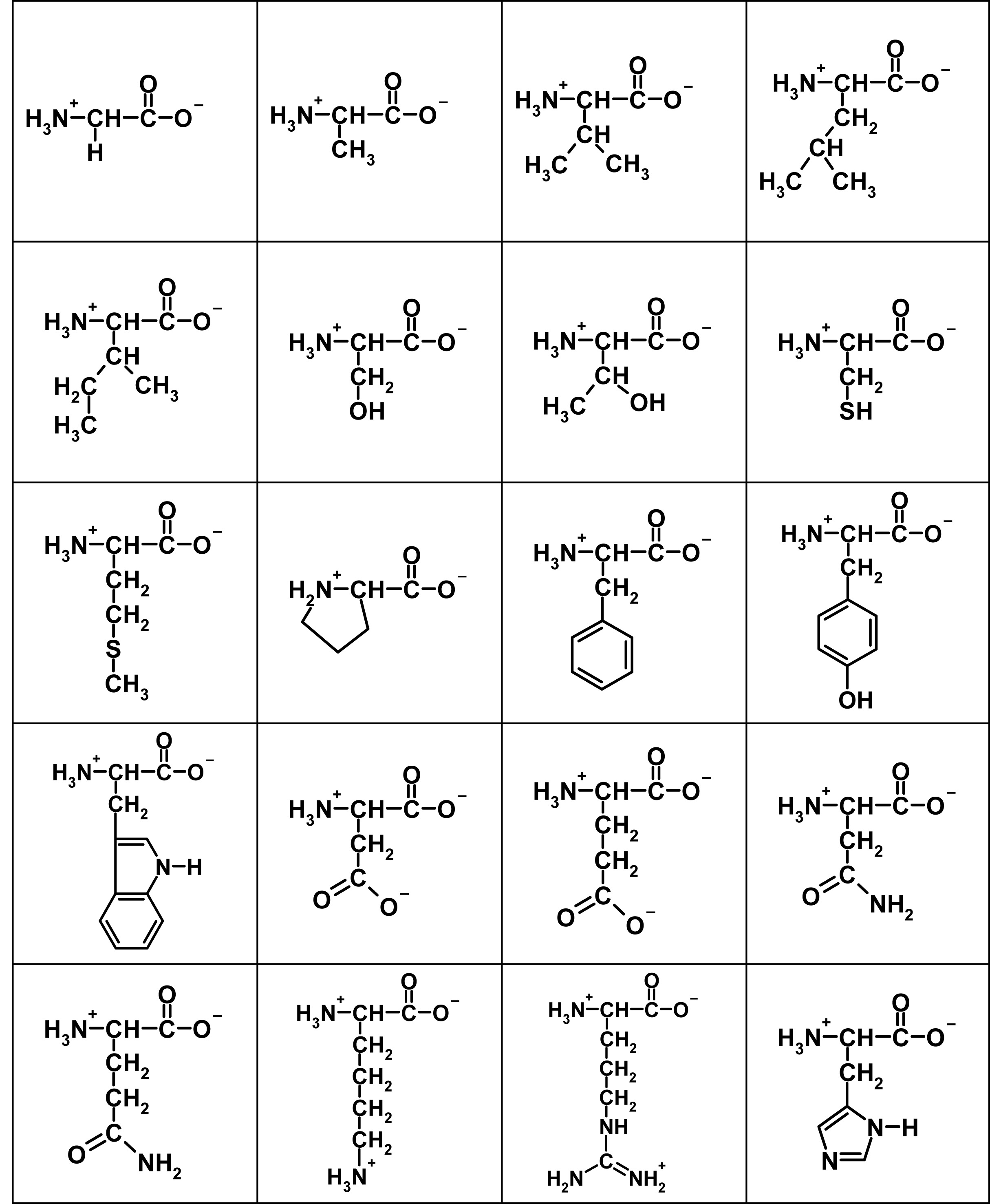
The salient features of these two-dimensional representations of 3D structure are the central (α) carbon with an H3N+– group to the left, a C-O– group to the right, and a side-chain depicted descending, with the simplest being a single hydrogen atom in Glycine top left. In a protein, these subunits are daisy-chained by linking the H3N+– group of one to the C-O– of another, eliminating two hydrogens and an oxygen (ie water) to form the peptide bond.
Because all acids are the L form – the D forms would have the side chain protruding upwards in the above chart – all side chains end up on the same side of the peptide-bond backbone. A freshly-synthesised peptide tends to shrivel up like a prude’s lips at gay Mardi Gras, as the various elements tussle for electrons with each other and with the surrounding water, and adopt the lowest-energy conformation. Like charges repel and unlike ones attract, while different side-groups have different affinity for the surrounding water and are variously repelled or attracted by it (hydrophobicity). The result is a complex fold adopted spontaneously and repeatably in the same physiological conditions. Where the peptide is an enzyme (not all peptides are), the fold brings the active site groups – usually just a handful of residues, often acting in tandem with a metal ion – into proximity, and the rest form a scaffold. The shape of this permits the entry of some substrates but excludes others, to give a degree of specificity, and its flexion can have a significant role in reaction kinetics.
Rather than name the acids, I will use a notation Row:Column to denote an acid by reference to the picture. Glycine, top left, is 1:1. Now, the substitutability of a given amino acid site depends very much on where it is, and what the substitution is. Substituting 1:3 with 1:4, for example, makes very little chemical difference. 1:4 is a little more hydrophobic (as this is not a strictly binary characteristic), and a little bigger (size, ditto!), so the minimum-energy conformation of the chain is slightly changed. But if the bulk of residues remain unchanged, the overall structure is much more constrained by that bulk than distorted by the substitution. Apart from a few key residues, such substitutions are unlikely to be catastrophic. But they do have the capacity to ‘tune’ the protein, in a beneficial or a detrimental direction, by the tiny steps postulated by the Darwinian mechanism. A poor fit can be progressively turned into a good fit, by blind ‘exploration’ of the protein neighbourhood. Other equivalent substitution groups can easily be identified from the chart – 2:2 for 2:3, 4:2 for 4:3, 5:2 for 5:3, even 1:2 for 2:1 etc. Substitution steps are not all equal in size.
Even though the sequential information can be rendered digitally, since it is modular, the mature folded peptide varies in an ‘analogue’ manner, by complex and continuous differentials between variants. There is no doubt that many substitutions are catastrophic, and many peptides cannot fold uniquely and repeatably, but this cannot be used to infer that all substitutions are doomed, nor that there is no way to gain a ‘toe-hold’ in the functional part of the space, which is essentially what Hoyle et al do. They assume, without evidence, that there is only one sequence in the whole of protein space that performs the function, and all the rest are duds. You can’t get there, and you can’t go anywhere else.
Any modern protein has been through the multi-generational filter of Natural Selection. It has likely explored its local protein space and this is the best it could come up with. While nearby variants may have been perfectly successful in their day, they cannot compete with the modern, ‘tuned’ version. So if they arise again, they are eliminated as detrimental mutations, where once they would have been top dog. This is a factor in the variation of both v and n. The smallest permutation space that includes a given modern protein includes all smaller spaces enfolded multiple times within it. Longer peptides appear to be closer to an optimum than shorter, but shorter strings drawn from a reduced amino acid set will still give peptides that perform biochemical functions. The range of functions, like the range of truly different acids, is limited. There are actually only about six. The enormous variety of the protein world comes from the wide variation of specificity of these fundamental reactions for different substrate molecules, controlled by the ‘wire frame’ shape and charge distribution. Even di- and tri-peptides (2 or 3 amino acids) can catalyse some of these basic reactions, though without the specificity of their giant cousins.
Nonetheless, protein space cannot be explored if it is not reasonably well-connected. One way to analyse its granularity with respect to function is to randomly sample a portion of it. It is necessary to restrict the search somewhat, because there are many more proteins that cannot fold reliably than can, but this is not the cheat it may appear. A 1993 paper by Kamtekar et al demonstrated a method of generating reliable α-helices (a very common motif in proteins) by simply dividing the amino acid set in a binary manner – polar and nonpolar – and creating a short, simple pattern based upon that found in natural protein helixes. The actual acid at any site was irrelevant, provided that it conformed to the appropriate polar/nonpolar nature of the natural residues. And these peptides folded. It is easy to see how a short stretch of such a fold – a dozen residues or so – can become a longer one, in a much bigger overall permutation space, simply by end-joining duplicates of the shorter sequences. And how such ‘modules’ can be moved around from protein to protein. All you actually need to gain a ‘toe-hold’ in the portion of the huge space occupied by working catalysts are one polar and one non-polar amino acid, in a pattern about a dozen residues long. 212=4096. Moving around that space is then a matter of various copy and paste mechanisms, not a fresh shake every time. Proteins are built from the substructures that work, not the many that don’t.
The extensive literature that cites the original Kamtekar paper, some shown on the same linked page, opens up a search space for the interested reader to explore the vast amount of work that has been done on the distribution of function in the world of randomly-generated, and randomly-swapped, peptide subsequences. The world of folded proteins is stuffed with function. This paper, for example, used a similar polar/nonpolar 14-acid patterning algorithm to generate a tiny portion of the space-of-all-peptides, which nonetheless contained functional analogues of 4 out of 27 natural peptides tried – a remarkable hit rate for essentially random sampling.
Hoyle’s imagined mechanism generates random, lengthy peptides from a raw amino acid mix. However, the probability of even a dipeptide in this ‘bumping into each other’ scenario is getting down towards zero. The ‘warm little pond’ is chemically naive; a strawman. Darwin (who coined the phrase) knew nothing of thermodynamics, nor protein. The free energy change associated with condensation/hydrolysis of the peptide bond means that it requires the input of energy to make it. The energy of motions of molecules in solution is not enough. Even with appropriate energy, having hit the jackpot once is insufficient. One has to retain that sequence, and this random process is not repeatable. So calculating ‘the probability of a protein’ by combinatorial means is irrelevant if that is not how it happened.
The problem does not go away, of course. We still have the thermodynamic and the repeat specification issues to contend with. A plausible solution to both is provided by the nucleic acids. It might appear that we have the same problem. Nucleic acid monomers need to polymerise, and for double helixes the specific ‘right-handed’ versions of the bases need to arise from a messier mix. This time, at least, the basic reaction is thermodynamically favoured. One of the nucleic acid monomers is ATP, the ‘energy currency’ of the cell. Although it is not a trivial matter to get the energy into ATP etc in the first place, once there, polymerisation can be driven by the available energy of the subunits. Besides their ‘energetic’ nature, an essential feature of the nucleic acid monomers is complementary pairing – A to T/U, and C to G. We could not realistically expect to start from a pure mix of right-handed monomers, neatly polymerising to form a single strand self-replicator to the exclusion of all contaminants. But this ability to complement, or hybridise, is a striking feature of nucleic acid strings. This isn’t just a mode of replication, but of stabilisation. Single strands will ‘fish’ their complement from a mixture and both strands are stabilised by the hydrogen bonding that ensues. Short chains made of consistently oriented subunits will complement more readily than those with mixed right- and left-handed monomers. In this way, non-replicating double helixes of complementary sequence would be the most stable form arising from a messier mix – a kind of purification process, ‘selecting’ those bases with optimal complementarity. Such structures cannot replicate, but this is a possible first step towards it without falling foul of the combinatorial issue – it is not necessary to locate a replicator sequence in search space before complementarity can evolve.
Like short peptides, short RNA and DNA strands have catalytic ability (ribozymes), and all the basic reactions are within their scope. One particularly relevant reaction is the ability to join an amino acid to a nucleic acid monomer, ATP, which can be accomplished by a ribozyme just 5 bases long. This is a central step in modern protein synthesis, the lone monomer now extended by an elaborate ‘tail’ arrangement – the tRNA molecule – and the joining now performed by a protein catalyst. Charging the acid in this way overcomes the thermodynamic barrier to peptide synthesis, because aminoacylated ATP has the energy to form a peptide bond where ‘bare’ amino acids do not. This gives an inkling of the mode by which Hoyle’s peptide space may have been actually accessed and explored. Short peptides formed by ribozymes from a limited acid library, with limited catalytic ability, may become longer and more specific and versatile by duplications and recombination of subunits. Meanwhile the acid library itself can extend by minor variations on the basic chemical themes until further variation is ultimately prevented by the extent of embedding of proteins in metabolism – it takes just one invariable site in an organism to freeze the underlying RNA codon assignment. There is a concomitant reduction of the catalytic role of RNA (though it remains, significantly, the catalyst for peptide bond formation). The one-handedness of the acid set would derive from the self-constraining one-handedness of nucleic acid monomers – because they are asymmetric, catalytic RNAs made from them are also asymmetric, and can only chain up one form of amino acid.
Allan Miller,
Thanks for engaging.
The interesting issue with ubiquitin is that it is part of a system that allows multicellular organisms to exist in both the fertilized egg form and the adult form where the cell cycle needs to be different or variable in order to accommodate both conditions and all in between. Just getting function of this system is not trivial and many proteins that were part of the prior system (prokaryotic cells) needed to be able to interface with this system.
When this systems performs is a suboptimal way disease is almost certain. Cancer is one example of a disease that is tied to the suboptimal performance of this system. Evidence also suggests the neurodegenerative diseases may also me tied to it. It occurs when instead of operating like a mature organisms cell the cell reverts to embryonic behavior.
I am very skeptical that there is a hill for this system to climb. A mind is possibility but it is a mind much more advanced than ours. The exciting part is we probably know less than 1% of having a complete understanding of how this system works.
Gil, and I received 2 objections to Gpuccios measurement method. The issue is if either of these objections scratched the surface of challenging the error factor gpuccio puts in his measurement. One was on a specific human protein mys 7 and other was Rums well articulated objection of the local optimum possibility. I did some homework on both and could not find any data that would suggest these challenge gpuccio. Any citing here would be great.
colewd,
Ubiquitin is present in free-living unicellular protists. The tagging of proteins by it is involved in all manner of cellular systems. Obsessing over its role in your favourite organism (the chimp!) is not relevant to its origination, which is what we really need to be talking of in the context of the OP. Its role in other systems has been co-opted for use in multicellular organisms.
Of course that can happen with Designs as well, but as it is evident that ubiquitin did not begin as a signalling system for animals, you are putting the cart before the horse in starting there.
We’d also need to wonder at the sequence conservation of multicellular ubiquitin-system proteins between protists and … um … chimps. That is certainly suggestive of common descent, while not conclusive.
Allan Miller,
I don’t think there is any identified difference in this system between us and chimps that has been identified at this point. The big transition is from unicellular and also from invertebrates. Gp[uccio mentions the inv to ver transition in the following chart.
https://uncommondescent.com/wp-content/uploads/2018/02/Ub3-1.jpg
Its interesting the information change in this system at this point of change from invertebrates to vertebrates.
Just claim again your disembodied magic mind did and and you’re done here. No need for that nasty evidence stuff.
Gosh. We’re pretty much the same, then? 🤔
My mention of chimps is a tease. Clearly, the only organism you really care about is humans. But when it comes to chimp/human divisions, it amuses me to pretend it’s because your real interest is the other organism in the pairing.
Large-step Information Change or lack of preservation of intermediate lineages? Failure to consider the latter cause of steps in data is a distinctive feature of your thinking.
Now what? Your interest has been piqued, what does the Intelligent Designer Investigator do next?
Allan Miller,
I don’t have a problem theologically with evolution. The issue is that the grand claims are not science. My real issue is with deception.
Well I can consider it all you want but right now were talking about the evidence we have. The real harsh reality to face is the simple to complex model is broken.
Correct. Thanks for the conversation. Happy New Year.
Only in your ignorance encrusted mind Bill.
You think I’m being deceitful? I might be inclined to invite you to fuck off.
Sez Bill. No it isn’t, sez pretty much everyone in academia.
That’s actually worth further comment. Basically you are saying that even if we lived on a planet where evolution was true, we’d have to conclude Design – something that didn’t happen – because evolution, by its very nature, sweeps up after itself, and creates ‘gaps’ in evidence.
I’d just like to add that there are prokaryotic homologues of ubiquitin. There IS evidence for the evolution of the eukaryotic ubiquitination system from a simpler prokaryotic one.
It may not be complete enough to reveal every aspect of that transition, but it does exist. Someone wrote an entire PhD thesis on that subject: The Prokaryotic Origins of the Ubiquitin-Proteasome System.
That thesis is from 2019, so it’s newer than Bill’s textbook, and Gpuccio’s blog post. It also has a lot more words and figures, and there’s no “labeling” in it. I guess it must be right then!
I think that Sal puts a lot more effort into his research than those arguing with him give him credit for.
For example Allan Miller wrote that what Sal does is to:
Even you in this reply wrote:
From his studies Sal interprets the evidence according to the logic of his world view. But you are no different. You (and Allan) also come to your conclusions according to the logic of your world views. You study variations between different genomes and conclude that the patterns of variety we see are due to past accidents and mistakes. This conclusion is not drawn from a knowledge of what actually happened in the past, it is an inference which fits in with with your beliefs. It confirms your world view to believe that genomic variety is the result of mistakes. The problem here is that by looking at what is happening at the genomic level through spans of time we can actually witness we see that genomes change for all sorts of reasons, some purposefully and some by outside influences none of which I would anthropomorphise as mistakes. The system has inbuilt features which control the amount of changes which will be passed on. You believe that changes slip through by mistake. But is it a mistake to allow a suitable amount of variation which would allow a species to adapt to an ever changing environment. Change is controlled by the system.
and:
Just because I believe that Sal has put some effort into trying to understand these processes does not mean that I think that those he is arguing against haven’t put in as much effort.
For what it’s worth I think that individuals from both sides are putting a great deal of effort in trying to bolster and confirm their particular world view.
That he puts effort in is not in dispute. But it all amounts to the same trick repeated. It impresses you, but then, you’re (broadly) on the same side. Anyone who puts a picture of a protein up and says ‘explain this!’ is a genius. 🙄
This, I have to say, is bullshit. I don’t have a prior ‘belief’ that it’s ‘accidents and mistakes’ (which I take to mean that mutations do not occur to serve a requirement), that is a conclusion from the evidence. I once had no idea how change occurred, and no strong need for it to be this way or that.
Do you think our ‘world view’ comes before or after our education? When I see an excess of silent over nonsilent substitution, or an excess of chemically conservative substitution, or an excess of transition over transversion, in genomic data, my understanding of the nuts and bolts of the processes leads me to conclude that this is good evidence that change is biased by chemistry, not by ‘need’. Design enthusiasts never offer reasons for these observed patterns. They may blink briefly, then carry on with their manifesto – “ahem! AS I WAS SAYING …”:
Where’s the evidence for this? You say it, it makes you feel warm and fuzzy (because of your ‘worldview’ objection to a process commencing with ‘mistakes’), but why should anyone buy it? What would distinguish a world with intent from one built entirely by cumulative selectable ‘mistake’?
Of course you do, of course. Keep telling yourself that lie, Charlie, if it makes you feel better
I think that is a very dismissive way of putting it.
Then you are gravely mistaken. Only one side here invests in shielding their particular “worldview” (spoiler: it’s creationism).
The other view represented here is the consensus of the global scientific community, which has procedures in place to purge personal bias, through hypothesis testing, experimental validation, replication of findings, sharing of data and methods, peer review and publication in professional journals.
I thoroughly reject your attempt to create symmetry here.
Hear hear! My 1-line post above yours originally had more substance in similar vein, before a ‘ah, what the hell’ moment caused me to take a razor to the draft!
Yes, I agree that Sal puts in a lot of effort. Unfortunately, it is wasted effort. He would do better to spend more time trying to understand why most biologists are evolutionists.
It has everything to do with life and evolution. Cancer is an example of unchecked positive feedback. Cells divide and multiply without any purpose outside of themselves and the more they divide the faster they multiply.
If during evolution an organism had appeared with equal capabilities to that of cancer cells within an organism then life would have quickly ceased.
We both agree that evolution is happening. What we disagree about is the role of accidents in the creation of novel forms and attributes.
I don’t have access to the full paper. They maybe are showing how a form of evolution happens in tightly controlled lab conditions, but how closely does it relate to actual life? How do organisms grow into new forms with new attributes, the latest being the ability for organisms to be consciously creative? How far does it go in answering this question?
Activities and processes such as is performed by ubiquitin in coordination with other molecular systems were present from the beginnings of eukaryote development. And this allowed life to evolve to the point where it could be aware of itself and its place in the cosmos. The whole is reflected in the parts. The single human zygote is a preparation for the self conscious adult. The primal eukaryotes had the potential to bring about the sophisticated nervous systems and sense organs of higher animals.
The individual life relates to earthly life as a whole in a way similar to how the parts relate to the whole in a hologram.
Allan Miller,
What does evolution is true mean?
Actually occurred – was in fact, rather than hypothetically, the reason for biological diversity.
I will pay attention to anyone who can help us to understand proteins. Protein complexes are not just static lumps of material passively floating around until they get incorporated into some structure or functional system. There are always new things to learn about protein complexes, their interactions and their functions. It doesn’t matter if that person has the knowledge directly or if they refer to where the knowledge can be found. I have found that sometimes the best teachers I have are those that I disagree with.
I didn’t say you had “a prior ‘belief’ that it’s ‘accidents and mistakes’ “, I said it was your conclusion.
Basically it boils down to a conflict between two opposing views, those who believe mind is primal and those who believe that matter is primal. I am one of the former and presumably you are one of the latter.
During.
I would hope we regard education as something that continues throughout life, and we are constantly refining our world view, and on rare occasions changing it.
These changes always involve chemistry. But the more we learn about the chemistry and the closer we look within organisms, the more intricate, interdependent, cooperative processes we find. There is always a struggle against external disruptive forces and sooner or later the organism dies. But organisms have very sophisticated ways of dealing with the various disrupting influences.
Just as individuals grow and develop in the direction of reaching their potential, so too life evolves in the direction of reaching its potential. Individuals reach their potential because inner systems differentiate and work together to maintain the whole. Likewise life on earth reaches its potential because organisms differentiate and work together to maintain the whole.
An example of control is the sophisticated DNA repair systems within organisms. Without these systems no organism would remain viable for long. But if these systems were one hundred percent successful then species would lose the variability that allows them to survive in an ever changing environment. Dynamic balance is called for and dynamic balance is what we observe.
dazz, Allan Miller, Corneel,
I’m not sure why you lot are getting so defensive. Looking for evidence to confirm our world view is something we all do whether we admit it or not.
That is exactly the kind of talk that winds people up. “I, Charlie, know better than you do yourself why you do what you do”. Channelling WJM and fmm. And, it’s wrong, as Corneel eloquently explained. So, doubly aggravating. This is why you get pushback.
But is everybody looking for evidence that could prove it wrong?
History tells us that the majority view is not always the correct view. If he is learning biology I don’t think his time is wasted.
I believe in evolution, but probably not in the way that most biologists believe in it. But I would not believe in something just because the majority believed in it.
Argument by analogy. The trajectory of individuals is a poor comparison to the trajectory of a genetic series passing through germlines.
There are mechanistic reasons why replication is imperfect. It would take far longer, for diminishing returns, if it were better. Also, perfection would be impossible to achieve by mutation, because the more perfect it got, the less mutation would be happening to make it better. See, I can explain the same thing, mechanically.
That’s not evidence of teleology.
What you’re really short of, though, is a mechanism. How does an anticipatory system operate? How does it know what to do, or predict the future? Does it make the same mutation in many individuals at once, or just in one and then arranges for them and their descendants to have a charmed life until fixation?
But he’s not learning biology. It’s noticeable because he misuses the vocabulary and fails to understand pretty basic stuff when things are explained to him. I have started by assuming that he knew what he was talking about only to see him miss things that a junior undergrad in biology would understand easily.
Agreed. But the problem here is that we’re not talking about just any majority. Biologists are constantly confronted by the evidence for evolution. Explanations in biology are enriched enormously once we take into account evolutionary relationships Evolution is not just a pervading paradigm, it’s a powerful one. It is so good that scientists tend to exaggerate on adaptive explanations (apologists take that as a sign of dishonesty or failure, but it’s the explanatory power of evolution that’s too blame for that tendency). Thus we have to be careful. But we have no option but to accept the evidence for what it is, because it’s so profoundly convincing and scientifically productive. That is why that guy said that nothing in biology makes sense except in the light of evolution.
ETA: The thread on the Copernican principle and Dark Energy is an example of poor thinking about why scientists hold to some paradigms. It’s not because we just want to, it’s because the paradigms help explain so many things. What J-Mac presented fails to explain a lot of the stuff, from large to minutia, that the current paradigms do explain. A new paradigm has to explain everything the older paradigm did and then some. It’s not enough to try and pseudo-explain a couple of things, no matter how huge or important they might sound.
That’s okay. I’m not here for your or anyone’s approval or agreement.
Corneel would have had a point if i had proclaimed that this is just an argument between scientists and fundamental creationists. But I didn’t make that narrow a distinction.
Theodosius Dobzhansky 😉
Good question. And that is only something each of us can answer for herself or himself.
Of course. Science would never progress if that weren’t true. The majority view was once 6 day Creation for example – a view Sal would like to see resurrected.
Me either. I’ve been wrestling for years with a non-mainstream view on sex evolution, for example. But it is worth stopping and wondering why a consensus is what it is. When was the last time an outsider made a significant contribution to a field? Einstein in 1905 is the only one I can call to mind.
Between scientists and pseudoscientists, then. Same applies.
You were mystified at our ‘defensiveness’. I was explaining it.
In science, other people have this annoying tendency to remind you about it.
I bet you recognize the type 😉
Argument from recognising the whole reflected in the parts.
I agree. That is a comparison of an observed process with with a reductionist abstraction.
I was comparing the observed appearance of differential forms at two levels.
CharlieM,
Oh, this one just struck me while I was walking the dog.
That’s a priceless example of proving precisely the point I was making! 🤣 “Chemistry you say? Oh yeah, chemistry. Anyhoo…”.
No, argument by analogy.
The passage of genes through the germline is an observed process. How did you get here?
Unjustifiably. What is true for individuals is not (necessarily) true for ‘life on earth’, organisationally speaking.
History also tells us that almost all apparently crackpot views turn out to be actually crackpot.
Neil Rickert,
What is the definition of a crackpot view.
Things completely unsupported by any evidence and contradicted by virtually every piece of evidence we do have, like
“a disembodied mind used magic to POOF biological life into existence.”
This is a really crucial point that’s easily overlooked. A successor theory needs to explain both (1) something that the previous theory couldn’t explain and also (2) why the previous theory seemed to have explanatory success that it did.
For example, general relativity is based on fundamentally different premises about space and time than classical mechanics — but one can show, using general relativity, why we would end up with the predictions of classical mechanics as long as objects are moving far slower than the speed of light.
It should also be stressed that not all theory change is a change in paradigm. (In fact the whole literature on paradigms is a mess — Kuhn doesn’t use the word consistently in Structure of Scientific Revolutions and subsequent developments in philosophy of science haven’t always helped.)
Who needs this kind of disembodied mind to use magic to poof life into existence?
The mindless disembodied substitute will do… as this comment proves that mindless, disembodied bolt of lightning can produce some biological life, unfortunately, without the mind… 😉
Sure thing…
I recommend a two-dimensional plot, the “Gruenberger-Baez” plot.
Note that high Baez scores and low Gruenberger scores indicate crackpottery.
True crackpots claw their way into the rarified region of the plot where Baez > Gruenberger.
Be wary of pulling the Fulton non-sequitur…
Sure, sure… as depicted above… 🙂
But note #13
There are no mechanical reasons why replication should happen in the first place. And even assuming replication just happened, why would it matter to any set of chemicals how long any activity was to take or what resulted from it?