
Simplistic combinatorial analyses are an honoured tradition in anti-evolutionary circles. Hoyle’s is the archetype of the combinatorial approach, and he gets a whole fallacy named after him for his trouble. The approach will be familiar – a string of length n composed of v different kinds of subunit is one point in a permutation space containing vn points in total. The chance of hitting any given sequence in one step, such as the one you have selected as ‘target’, is the reciprocal of that number. Exponentiation being the powerful tool it is, it takes only a little work with a calculator to assess the permutations available to the biological polymers DNA and protein and come up with some implausibly large numbers and conclude that Life – and, if you are feeling bold, evolution – is impossible.
Dryden, Thomson and White of Edinburgh University’s Chemistry department argue in this 2008 paper that not only is the combinatorial space of the canonical 20 L-acids much smaller than simplistically assumed, but more surprisingly, that it is sufficiently small to have been explored completely during the history of life on earth. The reason is that amino acids are not 20 completely different things, but a limited set of variants, each group of which varies mainly in shape and size, less so in chemical property. If one must use a textual analogy, this is not an alphabet of 20 different letters, but a half dozen or so, each of which can be rendered in several different fonts and pitches. But really, even roughly analogising them with language strings, and inferring restrictions on protein space from restrictions on locating or moving between viable strings in language space, fetches up against the rather obvious fact that amino acids are not letters, and proteins are not sentences or Shakespearean works.
There are obviously two parameters one can fiddle with to make the numbers look impressive – v and n.
- In the protein system in modern organisms, v=20 – there are 20 amino acids. Most proteins contain all 20. Where the asymmetry about the central (α) carbon atom permits the possibility of mirror image versions of the molecules, these occur invariably in their ‘left-handed’ version, but a ‘raw’ mixture generated non-biologically would give 39 different acids in all (one has no mirror image). There are even more possible acids, leading to a combinatorial explosion, in some imaginary ‘warm little pond’ with a mechanism gluing these things together at random
- As for n, the exponent in our space size parameter, it appears to be a feature of catalytic proteins (enzymes) that optimal structures are typically from several tens to thousands of subunits in length.
So – news to no-one – specific long proteins cannot be reached in a single step, especially if there are lots of different kinds of subunit to play with. A simple string of just 100 39-choice acids is one of 10159 permutations. If you think that’s still a bit cautious, stick the rest in. There are about 500 or so. 5001000? Too much even for Excel – the universe issues a #NUM!
Here are the flattened structures of the 20 amino acids:
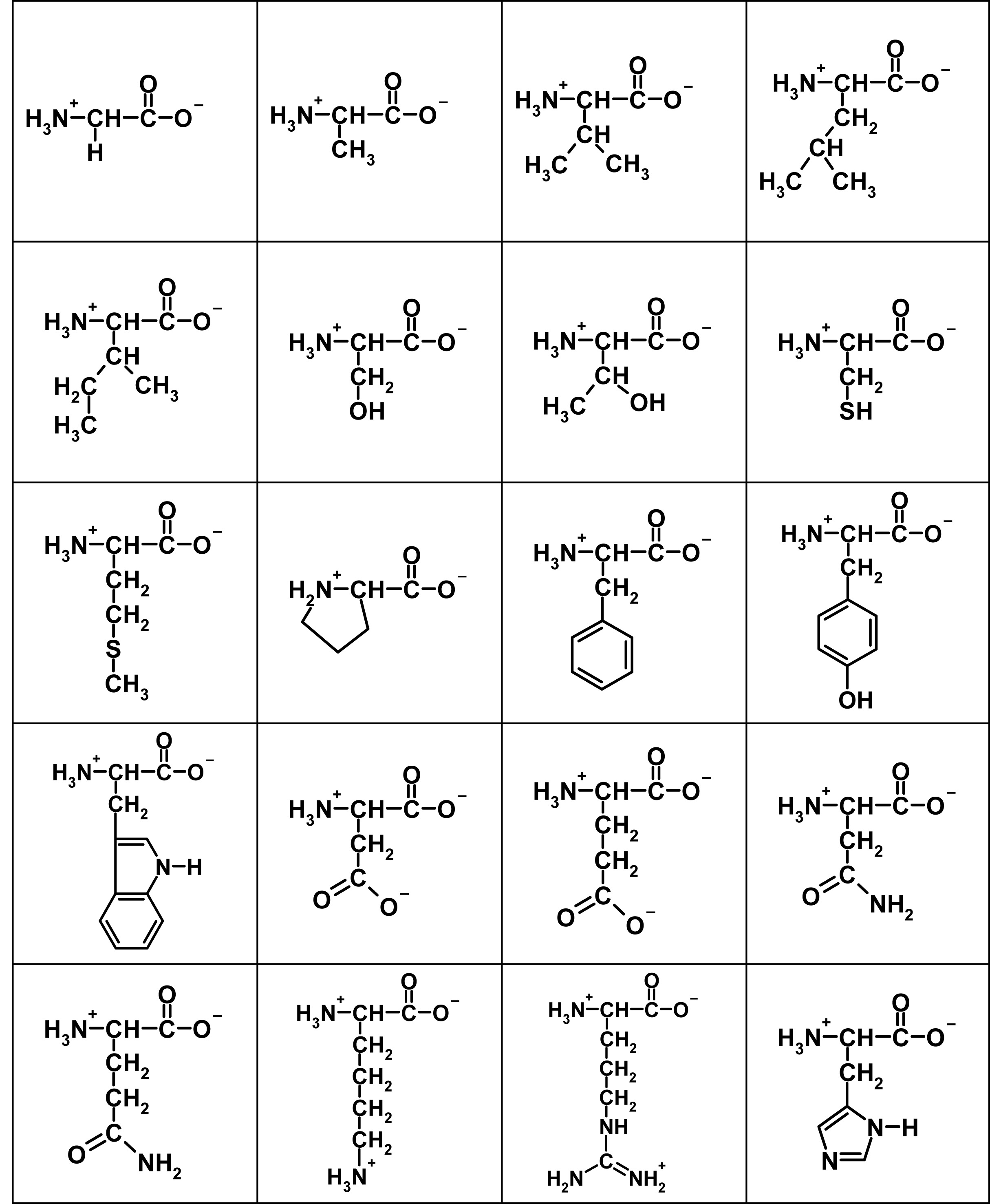
The salient features of these two-dimensional representations of 3D structure are the central (α) carbon with an H3N+– group to the left, a C-O– group to the right, and a side-chain depicted descending, with the simplest being a single hydrogen atom in Glycine top left. In a protein, these subunits are daisy-chained by linking the H3N+– group of one to the C-O– of another, eliminating two hydrogens and an oxygen (ie water) to form the peptide bond.
Because all acids are the L form – the D forms would have the side chain protruding upwards in the above chart – all side chains end up on the same side of the peptide-bond backbone. A freshly-synthesised peptide tends to shrivel up like a prude’s lips at gay Mardi Gras, as the various elements tussle for electrons with each other and with the surrounding water, and adopt the lowest-energy conformation. Like charges repel and unlike ones attract, while different side-groups have different affinity for the surrounding water and are variously repelled or attracted by it (hydrophobicity). The result is a complex fold adopted spontaneously and repeatably in the same physiological conditions. Where the peptide is an enzyme (not all peptides are), the fold brings the active site groups – usually just a handful of residues, often acting in tandem with a metal ion – into proximity, and the rest form a scaffold. The shape of this permits the entry of some substrates but excludes others, to give a degree of specificity, and its flexion can have a significant role in reaction kinetics.
Rather than name the acids, I will use a notation Row:Column to denote an acid by reference to the picture. Glycine, top left, is 1:1. Now, the substitutability of a given amino acid site depends very much on where it is, and what the substitution is. Substituting 1:3 with 1:4, for example, makes very little chemical difference. 1:4 is a little more hydrophobic (as this is not a strictly binary characteristic), and a little bigger (size, ditto!), so the minimum-energy conformation of the chain is slightly changed. But if the bulk of residues remain unchanged, the overall structure is much more constrained by that bulk than distorted by the substitution. Apart from a few key residues, such substitutions are unlikely to be catastrophic. But they do have the capacity to ‘tune’ the protein, in a beneficial or a detrimental direction, by the tiny steps postulated by the Darwinian mechanism. A poor fit can be progressively turned into a good fit, by blind ‘exploration’ of the protein neighbourhood. Other equivalent substitution groups can easily be identified from the chart – 2:2 for 2:3, 4:2 for 4:3, 5:2 for 5:3, even 1:2 for 2:1 etc. Substitution steps are not all equal in size.
Even though the sequential information can be rendered digitally, since it is modular, the mature folded peptide varies in an ‘analogue’ manner, by complex and continuous differentials between variants. There is no doubt that many substitutions are catastrophic, and many peptides cannot fold uniquely and repeatably, but this cannot be used to infer that all substitutions are doomed, nor that there is no way to gain a ‘toe-hold’ in the functional part of the space, which is essentially what Hoyle et al do. They assume, without evidence, that there is only one sequence in the whole of protein space that performs the function, and all the rest are duds. You can’t get there, and you can’t go anywhere else.
Any modern protein has been through the multi-generational filter of Natural Selection. It has likely explored its local protein space and this is the best it could come up with. While nearby variants may have been perfectly successful in their day, they cannot compete with the modern, ‘tuned’ version. So if they arise again, they are eliminated as detrimental mutations, where once they would have been top dog. This is a factor in the variation of both v and n. The smallest permutation space that includes a given modern protein includes all smaller spaces enfolded multiple times within it. Longer peptides appear to be closer to an optimum than shorter, but shorter strings drawn from a reduced amino acid set will still give peptides that perform biochemical functions. The range of functions, like the range of truly different acids, is limited. There are actually only about six. The enormous variety of the protein world comes from the wide variation of specificity of these fundamental reactions for different substrate molecules, controlled by the ‘wire frame’ shape and charge distribution. Even di- and tri-peptides (2 or 3 amino acids) can catalyse some of these basic reactions, though without the specificity of their giant cousins.
Nonetheless, protein space cannot be explored if it is not reasonably well-connected. One way to analyse its granularity with respect to function is to randomly sample a portion of it. It is necessary to restrict the search somewhat, because there are many more proteins that cannot fold reliably than can, but this is not the cheat it may appear. A 1993 paper by Kamtekar et al demonstrated a method of generating reliable α-helices (a very common motif in proteins) by simply dividing the amino acid set in a binary manner – polar and nonpolar – and creating a short, simple pattern based upon that found in natural protein helixes. The actual acid at any site was irrelevant, provided that it conformed to the appropriate polar/nonpolar nature of the natural residues. And these peptides folded. It is easy to see how a short stretch of such a fold – a dozen residues or so – can become a longer one, in a much bigger overall permutation space, simply by end-joining duplicates of the shorter sequences. And how such ‘modules’ can be moved around from protein to protein. All you actually need to gain a ‘toe-hold’ in the portion of the huge space occupied by working catalysts are one polar and one non-polar amino acid, in a pattern about a dozen residues long. 212=4096. Moving around that space is then a matter of various copy and paste mechanisms, not a fresh shake every time. Proteins are built from the substructures that work, not the many that don’t.
The extensive literature that cites the original Kamtekar paper, some shown on the same linked page, opens up a search space for the interested reader to explore the vast amount of work that has been done on the distribution of function in the world of randomly-generated, and randomly-swapped, peptide subsequences. The world of folded proteins is stuffed with function. This paper, for example, used a similar polar/nonpolar 14-acid patterning algorithm to generate a tiny portion of the space-of-all-peptides, which nonetheless contained functional analogues of 4 out of 27 natural peptides tried – a remarkable hit rate for essentially random sampling.
Hoyle’s imagined mechanism generates random, lengthy peptides from a raw amino acid mix. However, the probability of even a dipeptide in this ‘bumping into each other’ scenario is getting down towards zero. The ‘warm little pond’ is chemically naive; a strawman. Darwin (who coined the phrase) knew nothing of thermodynamics, nor protein. The free energy change associated with condensation/hydrolysis of the peptide bond means that it requires the input of energy to make it. The energy of motions of molecules in solution is not enough. Even with appropriate energy, having hit the jackpot once is insufficient. One has to retain that sequence, and this random process is not repeatable. So calculating ‘the probability of a protein’ by combinatorial means is irrelevant if that is not how it happened.
The problem does not go away, of course. We still have the thermodynamic and the repeat specification issues to contend with. A plausible solution to both is provided by the nucleic acids. It might appear that we have the same problem. Nucleic acid monomers need to polymerise, and for double helixes the specific ‘right-handed’ versions of the bases need to arise from a messier mix. This time, at least, the basic reaction is thermodynamically favoured. One of the nucleic acid monomers is ATP, the ‘energy currency’ of the cell. Although it is not a trivial matter to get the energy into ATP etc in the first place, once there, polymerisation can be driven by the available energy of the subunits. Besides their ‘energetic’ nature, an essential feature of the nucleic acid monomers is complementary pairing – A to T/U, and C to G. We could not realistically expect to start from a pure mix of right-handed monomers, neatly polymerising to form a single strand self-replicator to the exclusion of all contaminants. But this ability to complement, or hybridise, is a striking feature of nucleic acid strings. This isn’t just a mode of replication, but of stabilisation. Single strands will ‘fish’ their complement from a mixture and both strands are stabilised by the hydrogen bonding that ensues. Short chains made of consistently oriented subunits will complement more readily than those with mixed right- and left-handed monomers. In this way, non-replicating double helixes of complementary sequence would be the most stable form arising from a messier mix – a kind of purification process, ‘selecting’ those bases with optimal complementarity. Such structures cannot replicate, but this is a possible first step towards it without falling foul of the combinatorial issue – it is not necessary to locate a replicator sequence in search space before complementarity can evolve.
Like short peptides, short RNA and DNA strands have catalytic ability (ribozymes), and all the basic reactions are within their scope. One particularly relevant reaction is the ability to join an amino acid to a nucleic acid monomer, ATP, which can be accomplished by a ribozyme just 5 bases long. This is a central step in modern protein synthesis, the lone monomer now extended by an elaborate ‘tail’ arrangement – the tRNA molecule – and the joining now performed by a protein catalyst. Charging the acid in this way overcomes the thermodynamic barrier to peptide synthesis, because aminoacylated ATP has the energy to form a peptide bond where ‘bare’ amino acids do not. This gives an inkling of the mode by which Hoyle’s peptide space may have been actually accessed and explored. Short peptides formed by ribozymes from a limited acid library, with limited catalytic ability, may become longer and more specific and versatile by duplications and recombination of subunits. Meanwhile the acid library itself can extend by minor variations on the basic chemical themes until further variation is ultimately prevented by the extent of embedding of proteins in metabolism – it takes just one invariable site in an organism to freeze the underlying RNA codon assignment. There is a concomitant reduction of the catalytic role of RNA (though it remains, significantly, the catalyst for peptide bond formation). The one-handedness of the acid set would derive from the self-constraining one-handedness of nucleic acid monomers – because they are asymmetric, catalytic RNAs made from them are also asymmetric, and can only chain up one form of amino acid.
Not the one you would like to accept … since you don’t know the answer… Oh, well… So, what’s the point?
Yes, nobody says otherwise. It works or it doesn’t. But how well does it work?
You are either moving or you are standing still. There is no in between moving or standing still. There are, however, differences in how fast you move.
But biological functions do come in degrees. A molecule can bind another weakly, or strongly, and everything in between. It can catalyse the reaction at a very high speed, or slowly, and everything in between.
Or parts of it. You can recycle parts of a protein, and you can recycle them effectively (catch all nonfunctional versions and recycle them without any going to waste), or you can do it ineffectively (many are lost or missed by the system).
And that could be due to weak association between proteins. If that weak association is benefical, it can provide a basis for natural selection to enhance the function.
Once again we see the assertion with zero supporting evidence or argument. A vague general statement about what MUST be so. The viewpoint is just declared as if that establishes it as factual.
It does not.
It doesn’t matter whether you think I would accept it. If you have evidence that magic or miracles is required for new functions to evolve, present it.
But you don’t have such evidence, you’re bluffing and blustering.
Allan Miller,
This would be great if there was evidence supporting a smooth simple transitional structure starting with small peptides. The simplest observation is of greater then space shuttle complexity.
Knowing what we know now if there had never been a TOE such as universal common descent by natural selection do you think anyone would buy it as a new theory?
Both chicken and egg need to be there… and food.. (ATP)…
The miracle required is right in front of the once claiming to know, and yet… They say: “No no no… We do not accept this kind of miracle requiring a superior intelligence… We only accept the random explanation for the miracle; i. e. by sheer dumb luck… “
But random doesn’t cut it.
And?
Who can argue with this philosophical world view?
It’s pointless…
There is. And there are research groups studing that very question, such as the Tawfik group.
Here’s a recent paper on that:
Romero Romero ML, Yang F, Lin YR, Toth-Petroczy A, Berezovsky IN, Goncearenco A, Yang W, Wellner A, Kumar-Deshmukh F, Sharon M, Baker D, Varani G, Tawfik DS. Simple yet functional phosphate-loop proteins. Proc Natl Acad Sci U S A. 2018 Dec 18;115(51):E11943-E11950. doi: 10.1073/pnas.1812400115.
Meaningless rhetoric as usual. It’s not clear what “simplest observation” you are even referring to, nor what measure of complexity you’re using to compare biology to the space shuttle, nor do you at all articulate what the hell that has to do with evolution.
The case for it today would be even better, given what we know about DNA and how it evolves. The evidence for evolution has massively improved since Darwin’s day to such an extend that there is as universal a consensus as it is possible to get in any field of science.
As evolutionary biologist Eugene Koonin said in an interview:
“The word “discovery” may not apply quite directly here. It’s a transformation of the whole science, which is based on a variety of discoveries. The very approach to evolutionary studies has changed completely. Not only the fact of evolution itself but the existence of deep evolutionary connections between different domains of life — to be concrete — evolutionary connections between, let us say, mammals, such as humans, and prokaryotes, bacteria and archaea, have become indisputable.These findings make questioning not only the reality of evolution but the evolutionary unity of all life on earth completely ridiculous and outside of the field of rational discourse.“
Rumraket,
This is not true of eukaryotic control functions. A multicellular organisms cannot form without tightly integrated control functions containing millions of nucleotides. I takes very little to break these. The control functions are very complex. Plus complexity gets added all the time as we learn more.
Yes, DESIGN with a goal in mind is so much better than a random walk.
Eh, one doesn’t need the assumption of common descent to make sequence logo. Simply determining patterns of similarity and diversity is sufficient without a gratuitous token tribute to Darwin would suffice.
As I’ve said, the patterns of similarity and diversity are misinterpreted as common descent, when they are actually patterns that are optimized for scientific discovery, as evidenced by a logo that really doesn’t need the assumption of random mutation acting during common descent.
J-mac,
The role of QM that is well established is in the chemical covalent bonding in the primary structure of the proteins.
The other kinds of bonding and forces such as non-covalent bonds and hydrophobic “forces” (described by Albert’s textbook) are more classical physics as far as I know.
Silly person. You said that ‘quantum wibbling’ in DNA is involved in protein folding. But proteins – natural and synthetic – fold in the absence of DNA, ergo, yet another subject on which you are without clue.
But.. you know…evolution has to be gradual… So, the best way to ignore the facts that gradual evolution wouldn’t work in the vast majority of cases is it to ignore this fact and continue to ignore the evidence that contradicts one’s worldview…
Acknowledging the obvious is unthinkable… for some…
CharlieM,
You credit Sal putting deep thought into his post. I, presumably, just knocked mine up during coffee break. Wounded, I am! 🤣
That’s a false dichotomy. They can work not at all, badly, a bit better, well. You see an evolved function and insist it can only happen in 1 step. Why?
Haha, that’s your co-religionists thrown under the bus then!
Allan Miller,
In order to have variable cell division in multiple cellular animals you need very precise control that allow rapid cell division during embryo development or injury and stasis in a mature animal. This is a very complex and precise process. Jacks or better to open 🙂
The Darwinian paradigm does not make sense at the cellular level.
There is no more doubt about it. It’s fundamental quantum chemistry. But, there is more…
This may be true however, there is new research pointing to non-covalent quantum interactions in quantum chemistry.
” Found in translation: Quantum chemical tools for grasping non-covalent interactions
“This perspective overviewed the tools used to translate the highly mathematized quantum chemical description of non-covalent interactions into more intuitive concepts and graphical depictions. They function by extracting information from well-defined objects of the quantum physics world: the wavefunction, the energy operator (i.e., Hamiltonian), and the electron density. To a large degree, the strengths and weaknesses of an individual tool strongly depend upon which physical object is used in their construction. For example, wavefunction-based approaches are unlikely to break down for strong interactions, but, in contrast to Hamiltonian-based schemes (e.g., SAPT), they may lack the sensitivity to capture weak dispersion interactions. Density-based tools (i.e., QTAIM, DORI, and NCI), on the other hand, are often employed for visualization purposes…”
That’s hardly evidence against. After 4 billion years of ‘real’ evolution, it would be no surprise to find highly tuned structures. So we can’t say we’re not in such a world simply by pointing at the modern.
The counter proposal would carry more weight if someone could assemble a complex working cell from scratch. I’m inclined to think it can’t be done, for thermodynamic reasons. I think there has to be evolution from much simpler forms than we have available.
If Darwin hadn’t come up with it someone else would – indeed, others did. After years of fossil discoveries and the vast riches of molecular data, if it still hadn’t been proposed, I would be astonished. The evidence is vast. I know you can’t/won’t see it, but this is why it is rock solid in academia. I expect you’ve got you’re own theories about the latter too; I’m just telling it as I see it.
What’s that?
Your coffee spilling or your teeth grinding?
Either way… Your angered trolling is obvious…
Why do we need to consider multicellular animals at all? We’re talking about a very generalised argument here: protein space.
Of course it does. Most organisms are unicellular. They precisely serve the requirement of replication with variation.
Completely and utterly false.
Eukaryotic control functions are characterized by the fact that they come in degrees.
You confuse complexity with fragility.
I am shocked, shocked to find that…
Just a light-hearted expression of quantum uncertainties. Do you accept that proteins fold in the absence of DNA?
Allan Miller,
I agree. The problem is you have evidence of adaption but no evidence of transitions and the evidence so far shows large FI requirements for every major step. I think the design hypothesis is really surfacing here. It looks like it is rearing its ugly head in physics also.
You’re ignoring the point in contention, which was that Bill Cole asserted there is no evidence functional proteins can evolve from smaller peptides. Which there is, so Bill Cole’s claim that such evidence does not exist is wrong.
It’s ridiculous. How can something with even less to recommend it (taking your dubious assertions at face value) be ‘surfacing’?
“Too much FI so some bloke did it” 🤣
Allan Miller,
You have hit Dawkins argument. God is just too big a concept. My counter is the universe containing observers is too big a concept. Since we cannot seem to test the simple to complex model maybe its time to face a higher reality 🙂
One needs common descent to infer an ancestor. It’s not JUST a “sequence logo”. They are inferring a putative ancestor.
You seem to be implying they are just picking out some sort of chimeric sequence randomly from the similarities. Or that any putative chimera of extant sequences has the same odds of yielding a functional proteins. That’s not how it works as you can read from that wiki article on ancestral sequence reconstruction.
That does not make sense. They’re not “simply determining patterns of similarity and diversity”, which is meaninglessly vague gibberish.
They’re not just making an alignment and then picking out similarities or whatever nonsense it is you’re imagining.
They are constructing a phylogenetic tree and reconstructing what is inferred to be the ancestral form of the protein at the root node of the tree. That’s very different from just making some sort of chimera of extant sequences, which you mistakenly seem to be implying.
Despite having had it explained to you multiple times how and why that is wrong, you are still saying it, yes, but that doesn’t make it true. There is no other rational explanation for the existence of tree-structure in the data of shared similar protein(or DNA) sequences than the fact that they actually do derive from a common ancestor.
You once again don’t seem to understand the difference between sequences that are merely similar, and sequences that are similar and exhibit nesting hierarchical structure. But you can read about that here in Douglas Theobald’s 29+ Evidences for Macroevolution article.
Again, as explained to you many many times before over the last few yeas, in particular you need to understand the sections Prediction 1.2. A nested hierarchy of species, and Prediction 1.3: Consilience of independent phylogenies, which explain why your handwaving in the direction of mere similarities does not constitute a meaningful explanation for the manifest hierarchical structure in protein or DNA sequence data.
I do find a common issue with Creationists is an incapacity to think in the dimension of time. Everything is telescoped – at the extreme, into 6 days – in a habit cemented in early childhood. This seems to atrophy the capacity. So when presented with an alternative viewpoint that requires temporal thinking to grasp, it really does not compute.
Higher reality aka ‘bloke’. It’s not so much that God is too big a concept, it’s that it is, as an answer to the problems being posed, a dumb one.
The Design inference is a sham. It takes as a start point our ability to assemble within the restraints of physics, then performs a massive sleight of hand and infers an entity capable of transcending – indeed creating – the laws of physics. That seems unjustified.
Yes it is. There really is such a thing as degrees of binding, and differences in rate of catalysis, differences in the strength by which molecules bind each other, differences in the rate at which they diffuse or move throughout the cell, in eukaryotic organisms. You’re just brainlessly declaring something diametrically opposite to demonstrable fact. Once again.
It’s frankly ridiculous. One is not even required to really refute your posts, one can just point out the outright question begging fallacy every time. Please learn some critical thinking.
All mindless assertions that beg the question. This is it, this is your output. You just SAY these things and then appear to believe it is our job to prove it wrong otherwise we must believe it to be true by default.
How can you continue to fail to fathom that this isn’t how rational discourse works?
Rumraket,
How well do you understand Eukaryotic cell division and regulation? Degrees of binding are only the beginning of the issues. The function needs to work very precisely.
Rum you don’t appear to understand this subject. You are trying to cram stuff into an evolutionary paradigm that does not fit.
He’s certainly right about one thing: you work almost entirely by assertion. Why does it not fit? Bill Sez? First I’ve heard of it, at least.
Having had courses on it by cell biologists during my education, and by being employed at institutions doing biological research studying the phenotypic effects of mutations on cellular growth and metabolism(and what role these play in heritable human diseases), I know it well enough to know that what you’re saying is false.
Allan Miller,
Fair enough. Let’s start with this overview.
Aww, you’re so cute when you get condescending
No Bill, let’s not continue down every irrelevant rabbit hole you can think of to spam this thread with. There’s no reason to chase you around your latest Gish-gallop, playing whack-a-mole and nail-the-goalposts-in-place.
You make blind assertions diametrically opposite to demonstrable fact, evidence for that has been given now multiple times. The end.
Oh the irony!
Rumraket,
They are real facts you don’t want to face. There is no step by step process to a cell that requires protein recycling. If the recycling process falters disease is eminent. Gpuccio offers a very good overview here if you become curious. There are many papers in pub med showing how disfunction in this system causes disease.
Maybe if you copy-paste that sentence a few times into a couple of posts you will have magically made it true.
Rumraket,
The reality is until you understand the basics of ubiquitin system there is no way to go further in the discussion. If you have time read the article and we can discuss.
So the function is beneficial, and there are ways of reducing it’s efficacy. Hence if there are variations of the system that perform worse, there are selectable steps to it’s current function.
I’m sure there are. The function is beneficial, and many systems benefit from it. That doesn’t seem to be the kind of argument you think it is. In evolution, one entity can grow to become depend on a nother, such that if you remove it again, the loss is deleterious. That doesn’t mean the function didn’t or couldn’t evolve.
This is not a discussion, it’s just you declaring the same blind assertion over and over again, and numerous people pointing out that you are just committing the question begging fallacy.
Nonsense. If Gpuccio really thinks he has a point to make, why doesn’t he have the courage to publish, or at least engage with critics?
Bill may have forgotten!
déjà-vu on Ubiquitin
What do you mean “may have”? What has Bill NOT forgotten?
Bill doesn’t forget as much he flat out lies about things he’s been taught and shown simply because he doesn’t want them to be true. Bill is a Mighty Warrior For Jesus, so even if he tells the same lie about some aspect of evolutionary biology a hundred times God will forgive him. It’s not about scientific understanding with Bill, it’s about ensuring his ticket to heaven is punched.
Well, I’m prepared to stick a foot into the rabbit-hole, though I don’t know what would be served by that discussion. It’s all of the same character – here’s a modern protein, it couldn’t have got like that in one step, therefore ID. There are squillions of modern proteins to play that game with.
Ubiquitin is both troublesome and illustrative. Being … er … ubiquitous, it must (on the evolutionary paradigm) have evolved in an ancestor of the last Eukaryote common ancestor. So evolutionary history is scrubbed; evolutionary intermediates left no other descendants. That’s an issue – ‘how convenient’, the classically snotty online warrior may say – but it is a corollary of evolution that it tends to wipe history as it goes. That has to be borne in mind by anyone with a pretence to intellectual honesty in comparing paradigms.
The other issue with ubiquitin is its deep conservation. However, this doesn’t mean its sequence is functionally vital in all 70-odd residues. It needs an exposed lysine residue to support ‘chaining’. It has residues that can join to other lysines, to serine, cysteine, threonine and the N terminal amino group of the target. As to the rest, sequence is pinned in place by usage rather than function. I liken it the the ‘http://’ tag. That sequence is arbitrary, but so widely recognised that it becomes hard to vary without impact. A naive observer would consider it exquisitely designed for function!
Another thing to consider is variation. Although conserved, it does vary, and that variation follows the variation seen in all the other genes – there appears to be a different ‘design’ requirement for tagging in starfish, squid, dandelion and dry rot, which by remarkable coincidence follows that in their helicases, their spliceosomes, their rRNA etc. Almost as if there were Common Descent … 🤔
I don’t make that assumption, so though one might rightly fault them for such an assumption, I don’t make that assumption because I have better arguments!
It’s rather pointless to explore ALL of protein space once around when there needs to be several proteins matching each other to make complexes in systems all at once. It’s pointless no Helicase, or a helicase ring loader and ring breaker when there is no helicase! Or to make polymerase subunits when the other subunits are absent. And natural selection won’t work in such cases because the cellular system would be dead.
Sure, explore all protein space once, but how likely will that ensure several inter dependent proteins will emerge? And the OP assumes natural selection is available to help, which is a generous assumption if there isn’t anything alive to begin with.
You are just making the same argument though, merely extending the search space to include a composite string that includes both a protein and its interactor(s). Just as Hoyle, you ignore the possibility that the modern state can be reached incrementally. Why can’t an interaction be tuned by incremental evolution, such that the fit of the coupling is enhanced to the point of appearing deliberate, or at least remarkable in hindsight? You give no reason.
As I have said to you numerous times, I don’t favour a ‘proteins-first’ scenario. There are many reasons for that. As I’ve also admitted, there are problems with ‘RNA world’, but that would provide the answer to your conundrum as posed. After all, what would be the substrate of your helicases, in the absence of nucleic acids?