
‘vjtorley’ has honoured me with my very own OP at Uncommon Descent in response to my piece on Protein Space. I cannot, of course, respond over there (being so darned uncivil and all!), so I will put my response in this here bottle and hope that a favourable wind will drive it to vjt’s shores. It’s a bit long (and I’m sure not any the better for it…but I’m responding to vjt and his several sources … ! ;)).
“Build me a protein – no guidance allowed!”
The title is an apparent demand for a ‘proof of concept’, but it is beyond intelligence too at the moment, despite a working system we can reverse engineer (a luxury not available to Ye Olde Designer). Of course I haven’t solved the problem, which is why I haven’t dusted off a space on my piano for that Nobel Prize. But endless repetition of Hoyle’s Fallacy from multiple sources does not stop it being a fallacious argument.
Dr Torley bookends his post with a bit of misdirection. We get pictures of, respectively, a modern protein and a modern ribozyme. It has never been disputed that modern proteins and ribozymes are complex, and almost certainly not achievable in a single step. But
1) Is modern complexity relevant to abiogenesis?
2) Is modern complexity relevant to evolution?
Here are three more complex objects:

Circuit Board

Panda playing the flute

er … not yet in service!
Some of these objects were definitely designed. But all of them have undergone evolution. Nobody went up into their loft and came up with a complex circuit board prior to the first computer, or that – ahem – prototype jumbo prior to the first ever powered flight. A process of design resulting in any complex object invariably involves preliminary stages that are simpler. And yet this fact of design is disavowed when it comes to biology. It’s Design Jim, but not as we know it! Without a rationale other than modern complexities, we are informed that functional protein space starts at some threshold length and subunit variety beyond the reach of mere variation and assessment – the design methodology of trial and error, blindly applied in the case of ‘natural’ evolutionary processes. Somehow, a Designer is supposed to know, or find out by means unavailable to ‘nature’, where within this space function may be found. It’s conceivable, but if you disbar Chance on probabilistic grounds, Intelligence alone is no help without something to guide it to the target(s). If your first functional target is in the space [v=5, n=100], how do you find that out, and stop wasting your time in the lower-orders? And which of those 8 x 1069 sequences is it?
Torley makes a nod in the direction of Hoyle’s fallacy, and the Wiki that I also linked, but declares that such analyses “purport to take apart his argument without a proper understanding of the mathematical logic that underlies it.”. It is not clear what mathematical logic he has in mind, other than the generation of irrelevantly large permutation spaces by exponentiation. The mathematical relation involving string length (n) and subunit number (v) simply delineates the total space vn and the probability 1/vn of hitting a single unique target in that space if your string construction method involves draws of n instances of v variants. Beyond this, mathematics offers no further insight. The total size of the space matters only if it contains just one functional variant. Far more relevant to biology is the frequency of function in the space (and all its subspaces), its connectedness, the role of contingency, and the means by which strings are actually constructed (which does not involve hopeful lotteries from scratch).
The starting point of my post was this Dryden, Thompson and White (DTW) paper from 2008. I should make it clear that I do not regard the paper as beyond criticism, hence the “Journal Club” tag of my original post. I would take issue with a couple of assumptions
- They consider point mutation as the prime mechanism of string amendment, whereas recombinational methods broaden exploratory scope considerably.
- They expect Natural Selection to impose a greater barrier upon navigation than I think is warranted, as drift has a powerful effect on the ability of genotypes to break out of local adaptive peaks.
- They assume a vast starting population of unique sequences – ie, they choose a somewhat unrealistic model of evolution.
Since the evidence in fact points to a single common ancestor, LUCA, and a precedent lineage leading back to a first protein-coding organism, all evolutionary exploration must therefore be partly constrained by this ‘founder effect’. And indeed, there is likely a further founder effect dating right back from the members of LUCA’s presumably extensive protein repertoire to the first ‘coded’ gene. Subsequent catalytic peptides could all be, to some degree, highly scrambled derivatives of one or a few present in that original ancestor. Sequence originally not part of a coding gene may be incorporated, but the likeliest source of novel enzyme sequence is existing enzyme sequence, or fragments thereof. Since fragments can recombine, those that prove their worth in one catalyst will be those available for the modular construction of another. This is an important mechanism completely overlooked by Hoyle and his disciples. The text
“modulemodulemodulemodulemodule”
has a probability of 1 in 2730 = 8 x 1042 of occurring in a random set of 26-letter-plus-space picks, but “module” itself is only 1 in 3.8 x 106, with an obvious route to the longer string. And what do modern proteins most resemble – ‘modular’ structures, or completely random draws?
The relevance of synthetic peptides
That combination of modularity and random composition are the issues I attempted to highlight with my reference to this paper, one of my favourite examples of the functional richness of certain areas of protein space. In reassuring his constituency that the DTW paper has been thoroughly refuted (by repetitions of Hoyle’s Fallacy!), vjt omitted to discuss the profound implications of this study (it is but one of many in similar vein). I only mentioned it in passing, and have discussed it before, but it’s worth a deeper look.
This study was not an attempt to sample protein space per se, but to provide a method for constructing synthetic catalytic peptides using a combination of a ‘design’ heuristic and random variation within it. They examined only a minute subset of a subset of overall protein space, but despite that necessary restriction, their method included a substantial degree of random sampling of that local region – and happened upon function, by chance.
The authors created a library of ‘semi-random’ 102-amino-acid peptides. To apply the gee-whiz visualisation beloved of the Hoyle acolyte, a single example of every possible 102-acid sequence would require a mole of universes (6 x 1023) to contain them all. The authors’ subsetting technique resulted in a space with a mere 2 x 1053 possible members (the authors quote 5 x 1052, but I think they’ve miscounted one of the variants). This set of permutations would ‘only’ fill a sphere 4 AU across (twice the earth’s solar orbit in diameter) – big enough, but lost in that mole of universes. From this infinitesimal part of protein space, the authors created just 1.6 million examples – equivalent to using a discarded E. Coli shell as a scoop and filling it 1/10th full with proteins from the 4 AU sphere of permutations. And yet, within this tiny sample, they found functional analogues of 4 out of 27 E Coli knockout mutants assayed. That is, E Coli with disabled genes, unable to grow on medium deficient in particular nutrients, had their ability restored by a member of this tiny library. That is a quite remarkable hit rate. There is no particular reason to suppose that even one functional analogue of these modern proteins should exist in that entire 4 AU space – or in the many more spaces that could be sampled by similar means. Yet a scoop of proteins from this space invisible to the naked eye contains not one, but four functional proteins each suitable for a different task in a modern cell. If the sample is representative of the whole space, functional analogues of these 4 proteins alone would form a sphere 5 million miles across. On probabilistic reasoning, functional analogues of many of E Coli’s other 4,300 genes, as well as a substantial fraction of the genes of every organism on earth, are also highly likely to be well-represented in the basic 1.6-million-gene library, or at least the wider set from which it was sampled.
How much ‘random’ and how much ‘design’ went into the method? The authors restricted the space-of-all-peptides by use of a patterning algorithm known to produce folded catalytic peptides. The core element was a 14-acid pattern PNPPNNPPNPPNNP where P=polar residues (red) and N = nonpolar (yellow). This was bounded either side by fixed 4-acid units, and each ffff-PNPPNNPPNPPNNP-ffff helix was joined by a 4-acid ‘turn’ (blue), itself a random selection from a 9-acid group. The result typically folds into bundles of four alpha-helixes, individually one of the most basic of protein subunits.
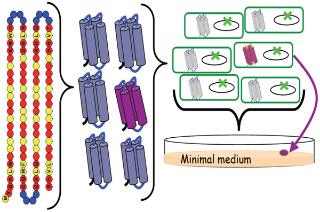
Here is the pattern, unravelled:
ffffffPNPPNNPPNPPNNPffffTTTTffffPNPPNNPPNPPNNPffffTTTTffffPNPPNNPPNPPNNPffffTTTTffffPNPPNNPPNPPNNPffff
The modularity of this should be immediately apparent. Even in 20-acid space, rather than Hoyle’s notion of a specific 102-acid structure arising with a probability of 1 in 5 x 10132, or even the chance 1 in 2.6 x 1080 of hitting a member of this patterned set by random picks of 102 acids, we could generate one of the polar/nonpolar chain elements with a probability of 1 in 1.6 x 108, the four-acid ‘turn’ sequence with a probability of 0.04, one of the 4-acid ‘fixed’ elements with a probability of 1 in 160,000. The probability of the complete structure arising, by stitching together such ‘modules’, is thus much greater than Hoyle would imagine. It does, of course, depend upon what the ‘subunit’ structures do – they must have some function individually, or in other combinations, to persist. But it should be clear that, provided a module can establish itself in one protein, it is readily available for extension and combination to generate the familiar modular elements that leap out from pictures of protein structure.
Dryden, Thompson and White
So, back to Torley’s critique, armed with a better handle on what modularity means for Hoyle-style probabilistic arguments. The bulk of his piece consists of proxy arguments against this ‘DTW’ paper which he claims ‘shoot it full of holes’. None appear to have been formally published, though perhaps by way of compensation, Dr Torley is careful to note the qualifications of his sources prior to presenting each. I will offer my own critique of their respective reasoning (which has a familiar ring).
1) Cornelius Hunter (blog posts).
Gets off to a fair start, by at least pointing out an evolutionary issue: the weakness of assuming a vast starting population of unique variants.
But…
The scientific fact is that the numbers are big. This isn’t a “game.” For instance, consider an example protein of 300 residues (many proteins are much longer than this). With 20 different amino acids to choose from […]
Bong! Hoyle’s Fallacy! Many modern proteins are longer than this. Modern proteins have 20 acids to choose from. Hunter pretends to make concessions by reducing v to 4 – but he sticks with n=300. Why? No sound rationale for this is given beyond the ‘modern protein’ gambit. His logic survives the apparent concession that 50% of sites could be unimportant – but he makes damned sure the rest are totally (and unrealistically) invariant. He comes up with 2 x 1090 (which is simply 4150). Again, a big so-what number, because he does nothing to ascertain that such an assembly with 150 invariant sites is the minimum functional linkage for a 4-acid library, nor that only a single target in that 4-acid 150-mer space has that function. It’s numeroproctology (H/T for that pithy bon mot to an unknown talk.origins commentator). Of course it’s a game. Pick a threshold – let’s say 1050 – and find numbers that exceed it. 2167. 3105 484. 572 […] 2039. Easy. Hunter doesn’t know what the minimal amino acid library or string length for functional peptides is, nor does he know the distribution of ‘function’ in the spaces delimited by those parameters (or any others). It’s just big and sparse, ‘cos Hunter sez so. As noted above, a tiny corner of 102-acid space has plenty of function suitable for the modern organism, contra Hunter’s 300-acid marker, and there is no reason to suppose it cannot be significantly smaller than this. The smallest catalytic peptide contains just two amino acids.
2) Douglas Axe (blog post).
The bigger problem, though, is that the DTW proposal also calls for radical reduction of the alphabet size. In fact, for this shortened sentence to meet their proposal, we would need to re-write it with a tiny alphabet of four or five symbols
I don’t think that Axe has grasped the paper’s argument with respect to the reduction of functional space due to common property. It is not the case that the authors assume a reduced alphabet (necessarily), but that even with a ‘complete’ alphabet of 20, many sites are capable of variation, either partial or complete, because most properties divide the set in a binary or ternary manner. As I have said, it is not so much different ‘letters’, but the same letters rendered in different fonts. So if a site requires only that ‘an amino acid’ be present, 20 ‘different’ sentences represent the same thing – aaaaaXaa, where a is constrained but X could be anything. Repetition is redundant; they are all the same functional ‘target’. The specificity at other sites requires only that the acid be hydrophilic or hydrophobic – 20 ‘variants’ can thus be represented here by two: aaaaaYaa and aaaaaNaa.
Nonetheless, although DTW may not be formally proposing a primitive reduced acid alphabet, many do (and I include myself). So what of Axe’s claim that shortening sentences and reducing alphabets reduces ‘meaning’ (in his strained analogy with text)? Simply: he’s going the wrong way! ‘Primitive’ proteins may not possess anything like the elaboration of modern forms, but they only have to be better than what else is around at the time. You don’t go there from here, you get here from there. Insisting on a lower limit is like suggesting that a primitive language with only one syllable cannot be functional. You could not do Shakespeare justice with it, but “ah!”, “ah?” and “aaaaaaarrrrggghhh!” could still come in handy!
Then, despite a wealth of references in the DTW paper to evidence that protein neighbourhoods are predominately functional, Axe declares the opposite conclusion:
We know that amino acid changes tend to be functionally disruptive even when the replacements are similar to the originals
Axe links a reference to his own work to back up this claim – a paper upon limits to the serial substitution of outer acids upon protein function. This paper does not justify the conclusion that ‘changes tend to be functionally disruptive’. ‘Concentrating one’s fire’ on exterior sites is not an approach that is taken by evolution, and so cannot be used to draw general conclusions about it. The untranslated DNA sequence has no information about where in the final protein the codon’s acid will end up – all sites are impartially available for mutation, and each change affects the landscape. It is not possible to rule out an evolutionary path to a modern protein unless you have tried all the possibilities (which are not limited to point mutation).
In fact, one of the IDist’s favourite facts, the error-tolerance of the genetic code, depends entirely upon protein neighbourhoods being ‘probe-able’, counter to Axe’s declaration. The arrangement of the genetic code means that the likeliest translation errors tend to result in substitution by a member of the same acid property group. For this to have any physiological significance, it must be the case that these substitutions still result in a working protein. Therefore, the same change permanently made, by a mutation in the DNA, must also lead to that working protein. Further support is lent by the existence of polymorphisms in populations, sequence divergence among higher taxa, alternative splicing, and a wealth of literature where chopping protein up in all manner of ways yields functional products.
Torley abstracts two of Axe’s questions, to which I offer answers:
Q: After all, why would cells go to so much trouble making all twenty amino acids if far fewer would do?
A: Because proteins made of 20 amino acids are capable of more subtle variation than those made of a few. This does not mean that few-acid proteins cannot function, but that there is selection for more finely-tuned products (which appears to also select for increase in length). Consider a symbol set consisting of only vertical and horizontal lines. Crude drawings would nonetheless be possible, but adding diagonals and dots and curves and circles improves the palette greatly.
Q: And if fewer really would do, why do cells so meticulously avoid mistaking any of the twenty for any other in their manufacture of proteins?
A: Because in some protein somewhere, in any modern cell, every one of those acids has come to occupy a vital position. It does not have to matter everywhere for the mechanism of identification to matter somewhere, and therefore for accuracy to be vital throughout.
3). Kirk Durston (letter, no online source)
My concern has to do with whether functional sequence space shrinks to zero before the size of the total sequence space becomes small enough to be adequately explored.
Or, putting the evolutionary horse back in front of the cart, the concern is that an amino acid library up to a certain threshold can create no string of any length that is functional. Which is unsupported. What function does one have in mind? Catalysis? True, short monotonous peptide catalysts are less specific and kinetically subtle than their modern cousins. But it is wrong to say that therefore they must be useless. Additionally, catalysis is not the sole role of protein. They can function as structural elements. A polymer made of just one acid, Glycine, for example, forms sheets, so while catalysis may be beyond the reach of a single-acid peptide bonding mechanism, there is still a potential functional structural role. The subsequent addition of a second or third acid can start to take one into the region occupied by catalysts. The library can expand by codon group subdivision until every acid has individually assumed a critical role in one or more proteins, at which point further expansion or substitution is severely constrained.
Durston then discusses the experimental difficulties associated with reducing to a binary code for a universal ribosomal protein. However, since modern organisms (and presumably LUCA) possess ‘tuned’ 20-acid proteins and ribosomal RNA, each evolved in the presence of the other, it is not reasonable to expect to be able to reverse evolutionary history simply by fiddling with one element of the complex. Cars don’t run well on cartwheels either.
Torley describes Durston’s logic as ‘skilled’ and ‘rapier-like’. That is, putting it mildly, over-egging it. 🙂
4. Branko Kozulic (publication details unknown – the link is a PDF)
In [the] laboratory, researchers have designed functional proteins with fewer than 20 amino acids [10, 11], but in nature all living organisms studied thus far, from bacteria to man, use all 20 amino acids to build their proteins. Therefore, the conclusions based on the calculations that rely on fewer than 20 amino acids are irrelevant in biology.
This is not the case. The fact that no organism today uses fewer than 20 amino acids tells us that either
a) No organism ever existed with fewer than 20 amino acids.
b) All lineages that used fewer than 20 amino acids have become extinct (apart from those that ultimately gave rise to LUCA’s 20-amino-acid library).
Kozulic nails his colours firmly to mast a), but gives no reason to discard b). Positive reasons to consider b) include the evolutionary relatedness of aminoacyl-tRNA synthetases and tRNAs and the relationships among the amino-acid biosynthetic pathways. Additionally, the ‘error-tolerant’ codon grouping pattern is consistent with b). Expanding the acid set necessarily means subdividing parts of the 64-codon set. This is constrained – not all subdivisions are of equal chemical impact. What is true for minimising point error in one protein is true for genome-wide substitution: the likeliest global addition to a codon group is an acid with similar properties, automatically ‘error-tolerant’ in translation for the same reason.
Concerning protein length, the reported median lengths of bacterial and eukaryotic proteins are 267 and 361 amino acids, respectively [12]. Furthermore, about 30% of proteins in eukaryotes have more than 500 amino acids, while about 7% of them have more than 1,000 amino acids [13]. The largest known protein, titin, is built of more than 30,000 amino acids [14]. Only such experimentally found values for L are meaningful for calculating the real size of the protein sequence space, which thus corresponds to a median figure of 10^347 (20^267) for bacterial, and 10^470 (20^361) for eukaryotic proteins.
The total size of the space-of-all-real-proteins is obviously delimited by the size and composition of the largest protein at that moment in time. So what? What was the largest protein 3 billion years ago? 3.5 billion years ago? 3.8 billion years ago? These, and the distribution of function in the corresponding space, are the parameters of relevance to the probabilistic viewpoint. I don’t know, but then, you don’t either. It’s not clear why Kozulic thinks bumping up the figures is relevant. The argument ‘you can’t have little proteins because there are big ones’ is obviously bogus; the alternative: ‘you can’t get big proteins from little ones’ is little better.
RNA
Finally (thank heaven!) we move onto the RNA world. Torley claims that I make a ‘concession’ with regard to thermodynamic difficulties with prebiotic protein synthesis. My only concession is to the laws of physics, particularly the 2nd law of thermodynamics (the real one, not that silly attempt to bring Shannon entropy into biology). I’m surprised he’s surprised – all scientists try to accommodate facts! But on the thermodynamic issue, RNA monomers carry the energy to drive their own polymerisation, which is a significant strike in favour of the RNA world. Amino acids need to be activated to cross the thermodynamic barrier. But rather handily, that activation can be achieved by ATP, an RNA monomer.
It is by no means a universally accepted idea, however. Larry Moran at Sandwalk, for example, is a trenchant critic in the blogosphere, and some of the reviewers of this nice paper by Harold Bernhardt (check the title!) pull no punches in showing their dislike of RNA-based speculation. But it remains my favoured bet, with significant evolutionary, energetic and (with some caveats) chemical logic. Torley counters by showing a picture of a ribozyme. Complex, huh? Yet, as Bernhardt notes, the smallest known ribozyme contains just 5 bases. It aminoacylates ATP – that is, it ‘activates’ the amino acid, the very thing needed to escort peptide synthesis in a thermodynamically favourable direction noted above. Putting RNA first thus gives both energy and specification – a nod to both ‘true’ and ‘faux‘ thermodynamics!
Bernhardt also discuss the Harish & Caetano-Anollés paper that Torley links (via ENV) in the belief that it dismantles the RNA hypothesis due to a possible early link between protein and the (RNA) ribosomes that make it. I find this paper being pushed across the virtual desk at me increasingly; it has obviously found its way into the clip-basket of many a dogged anti-evolutionist, without, I think, a full understanding of the issues. It is particularly amusing that the methods used to determine the depth of this link between RNA and protein rely entirely upon Common Descent. Ribosomal proteins and RNA are mined for divergence signals across kingdoms. I don’t know where Torley stands on common descent, but more than one poster I’ve seen bigging this paper up also thinks phylogenetic analysis is hogwash … don’t buy the methods; love the conclusion!
Be that as it may: the conclusion that this is the death-knell of the RNA world is wholly unfounded. It derives from some poor reporting and understanding. The issues are discussed more fully in the Bernhardt paper above (see section “Proteins first”), but fundamentally, the proteins analysed were all synthesised in ribosomes, from mRNA copied from DNA. In this, they are no different from any other modern protein. You cannot conclude from phylogenetic analysis that they are as old or older than the ribosomes that make them, since the signal of the protein phylogeny is rooted in ribosome-processed DNA. Although proteins may in fact have been involved in the early RNA world, they weren’t these proteins – ‘genetic-code’ proteins, ribosomally synthesised. The simple explanation of ribosomal protein is that, once protein started to be synthesised, it found uses throughout the cell, including many components of the protein synthesis system.
And regardless, you can’t extend ribosomal data to dismantle the whole of ‘RNA world’. However tightly coupled the ribosome (the site of protein synthesis) may have become with protein, even early in its evolutionary history, this forces no such constraint on any other ribozyme (any nucleic acid catalyst).
Could someone please put in a “fold” so that Alan’s post “continues below the fold”? It’s just pushed all other posts off the bottom of my browser.
Done 🙂
Drat! I knew there was something I’d forgotten before pushing ‘publish’!
Thanks very much for this post, Allan. I was going to do an OP on the Harish &
Caetano-Anollés paper (which I got not from an ID link but from a Google Scholar search for ribosome evolution!) but this is much better.
Lizzie,
I really like the Harish/Caetano-Anollés paper – it’s a neat attempt to reconstruct history. But while phylogenetic methods may be fair for reconstructing relative sequence of events, I think Caetano-Anollés himself may have over-stretched the interpretation of the results. Dated molecular phylogenies typically need recalibrating according to fossils, as they drift apart for various reasons, but obviously that is not possible with some of this data. Deep branches are a notoriously poor fit.
And when you start comparing DNA divergence via what are effectively three different macromolecules – RNA, structural protein and catalytic protein – you can’t expect evolutionary rates to map across them readily, because different constraints apply. I’m sure that the authors are better qualified than I am to judge these matters, but these difficulties with absolute dating inform my reservations over interpretation. And the fundamental blocker as to whether this provides a window on the ‘RNA world’ remains: none of these structures is a direct product of the RNA world at all, but of the ‘DNA world’. While ribosomal sequence may descend (via DNA) from primitive RNA structures, preserving functional equivalence, protein-coding sequence has no equivalent ‘meaning’ outside of the translation system. That’s as far back as it can probe, to its own origin.
I could have saved myself a lot of time. Here’s vjt, in an OP directed at Lizzie’s ‘CSI’ picture, but referencing his own posts on Protein Space:
If you can’t actually find a smaller replicator, the smallest possible replicator is the smallest one actually replicatin’ – a 4000-gene ribosome-totin’ modern cell! If ‘Darwinists’ can’t find anything smaller than that, there wasn’t one. Ever. There was, instead, this ‘protein fairy’ … 😉
http://profiles.nlm.nih.gov/ps/retrieve/Narrative/PX/p-nid/197
Vincent Torley has acknowledged Allan’s post as:
You’d be very welcome to pursue issues here, Vincent.
Yes indeed 🙂
gpuccio seems to have taken on the task. See post #99 at http://www.uncommondescent.com/intelligent-design/csi-revisited/#comment-455060. Any comments?
Looks like I’ve missed my chance for anyone to see my post…so please forgive me if I cut and paste this into a later discussion on the same topic!
You’ve pointed out that the proteins in primitive or quasi-life probably worked far less efficiently than proteins in todays organisms. It seems to me that makes Fisher et al.’s result even more promising. They got 4 functions out of 10*6 proteins assaying for 27 functions. But how many proteins did they miss because the protein had only 1/100th of the function necessary to rescue EColi? That little function doesn’t cut it in the mega-efficient streamlines biochemisty of an EColi but a few billion years ago it was the only game in town. It would be like trying to rescue ‘flight’ in an F-22 fighter with the Wright Brothers engine from Kitty Hawk.
RodWilson
Hi, Rod and welcome to TSZ. First comments from new subscribers need to be approved but thereafter your comments will appear immediately.
underthetable,
Coming very late to this!
He airily dismisses the RNA hypothesis, without really coming to grips with the rationale. That short RNAs are catalytic is dismissed with “so what”. How does one deal with such a critic, when the whole discussion is about combinatorial arguments? Sheesh. Because we have no RNA organisms, there was no RNA world, and a Designer must have made LUCA ‘cos there’s no other way, seems to be the thrust of it. Meh.
I anticipated the “the proteins were designed!” attack even as I wrote, and covered it in the text. The basic ‘designed’ pattern was 14 residues in length, well within the reach of a random process itself. Variation within that constrained pattern was completely random. The random evolution of a 14-acid binary pattern is trivial, as is the stitching of such modules into longer repeats.
They have to take some short cuts – what are people expected to do? Construct totally random peptides with no context and throw them at .. what, exactly? The proteins were designed only inasmuch as the 14-bit-binary-plus-turns pattern was known to produce a fold, and folding matters.
The paper demonstrates that many different functional proteins can be randomly generated by shaking a corner of protein space likely to contain functional proteins, and that that corner is plausibly accessible without invoking a ‘UPB’. It’s not looking for function throughout protein space. It is one of many such studies, and along with mistranslation, polymorphism, sequence variation across taxa, alternative splicing, protein family homologies and the possibility that noncatalytic function preceded catalytic, gives the lie to the Creationist assertion that protein function is and ever was sparse, and the Hoyle-inspired error that it is the total length of the peptide that matters, not subunit combination.
The lack of detectable catalytic activity in an assay for the original protein is, I admit, a valid criticism of a strict like-for-like replacement hypothesis. Further work would need to establish the mechanism of replacement. But if a (partially) random, completely unrelated peptide can rescue random functions, if badly, that means that living peptides are replaceable, and potential ‘targets’ common. gpuccio can’t see the significance of this for an evolutionary model. I don’t know why.
Alan Fox,
(Philosopher) VJ loves a qualification-fest, doesn’t he? 😉
RodW,
Sorry, completely missed this too!
Yes, I think you’ve hit the nail on the head there. You can do things badly if nothing else is doing it better.
Also, I made mention of this, but essentially, both the proteins and the functions were chosen at random (with a caveat regarding the acknowledged design of the patterning algorithm). They chose E Coli knockouts for availability and convenience of experimental assay. They could equally have chosen any protein in any organism ever, but for obvious experimental difficulties with blue whales, T. Rex and such. It’s the intersect of a tiny random sample of proteins and a tiny random sample of replacement functions which makes this (potentially) so significant in the context of ‘improbability analyses’.
But I acknowledge, as an issue requiring further work, the unknown mechanism of replacement, pointed out by gpuccio.
Just a general observation about the silliness of the ID/creationist N^L calculations;
This year’s Nobel Prize in chemistry went to Martin Karplus, Michael Levitt, and Arieh Warshel for computer simulations of biomolecules.
There is a rather nice summary of their work and its history on pages 13-16 of the December 2013 issue of Physics Today.
The history of this field goes back to 1967 and much before; and they certainly don’t calculate everything using N^L.
One can pick up any number of introductory statistical mechanics or quantum mechanics textbooks and quickly learn that chemical bonds are not all equal and that they are temperature dependent. In fact, even high school chemistry teaches about the various types of bonds that occur in complex molecules. The calculation N^L has no temperature dependence, and it is simply irrelevant to any assembly of atoms and molecules into a complex biomolecule.
With even a little knowledge of chemistry and physics, one can quickly reject any ID/creationist “calculations” and assertions about complex molecules. Chemists and physicists now understand that complex molecules evolve and don’t come together in a flash in a warm little pond somewhere.
Mike Elzinga,
It is certainly frustrating to go into detail over the essentially non-digital nature of active biopolymers – of phenotype – and find digital arguments returned by way of refutation!