
Evolution of KRAB Zinc Finger Proteins vs. the Law of Large Numbers
There are patterns in biology that violate the law of large numbers, and thus suggests Intelligent Design or at the very least statistical miracles. The pattern involves KRAB-ZnF proteins that have multiple zinc finger domains side by side that are inexact copies of each other and would require a scenario of co-evolution of their DNA binding partners with every additional zinc-finger insertion — a scenario indistinguishable from a miracle.
The role of a zinc finger can be something like a clamp or a lock. Each zinc finger in a KRAB-ZnF protein is fine-tuned to connect with a DNA much like a lock (the zinc finger) can receive a key (like DNA). Here is a depiction of a KRAB-ZnF protein with 4 zinc fingers as part of a large chromatin modifying complex. The four zinc fingers are marked “ZN” and connect to DNA:
The evolutionary explanation of side-by-side repeated patterns of zinc fingers such as in KRAB-ZnF proteins shows a severe lack of critical thinking by evolutionary biologists who pretend “phylogenetic methods” are adequate explanations of mechanical feasibility of common descent.
To illustrate the problem, consider the KRAB-ZnF protein known as ZNF136. For reference, this is the amino acid sequence of ZNF136:
https://www.uniprot.org/uniprot/P52737.fasta
This is an amino acid fragment contained within the ZNF136 protein
TGEKLYDCKECGKTFFSLKRIRRHIITH
This short sequence is called a zinc finger domain which in 3D looks like this:
https://sciencescienceeverywhere.files.wordpress.com/2015/08/figure-11.jpg
{kind=link}
Wiki gives a description of the function of zinc fingers in proteins that have them:
http://wikipedia.org/wiki/Zinc_finger
A zinc finger domain requires two “C” amino acids and two “H” amino acids placed in the right positions. It requires a few other things too…
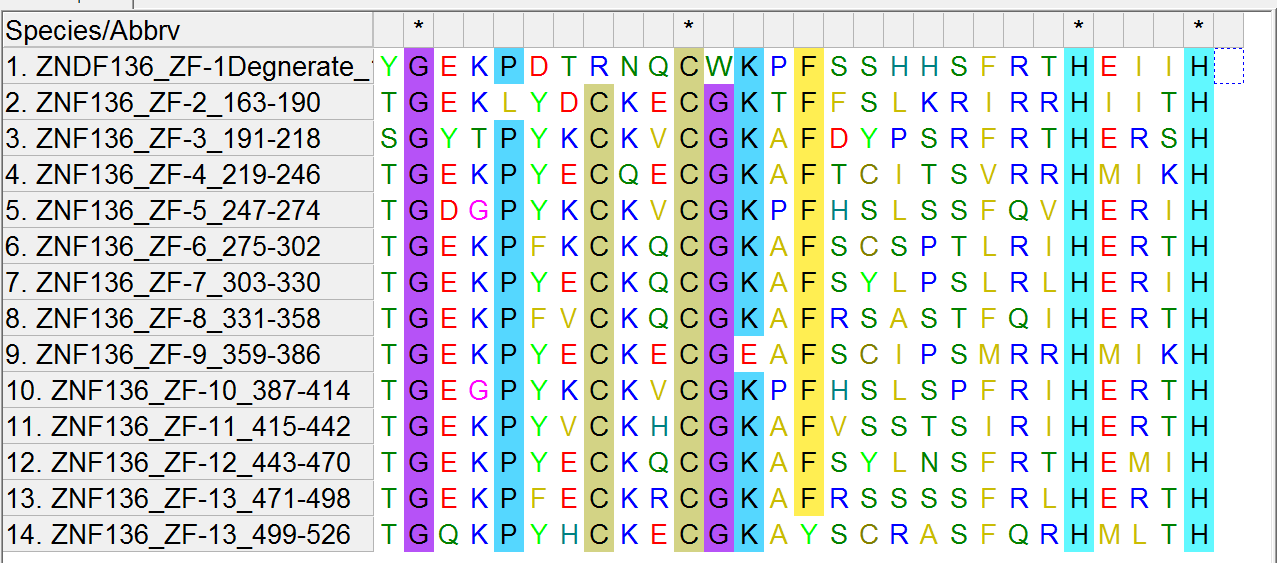
There are 13 zinc fingers in the ZNF136 protein and these are their sequences:
TGEKLYDCKECGKTFFSLKRIRRHIITH
SGYTPYKCKVCGKAFDYPSRFRTHERSH
TGEKPYECQECGKAFTCITSVRRHMIKH
TGDGPYKCKVCGKPFHSLSSFQVHERIH
TGEKPFKCKQCGKAFSCSPTLRIHERTH
TGEKPYECKQCGKAFSYLPSLRLHERIH
TGEKPFVCKQCGKAFRSASTFQIHERTH
TGEKPYECKECGEAFSCIPSMRRHMIKH
TGEGPYKCKVCGKPFHSLSPFRIHERTH
TGEKPYVCKHCGKAFVSSTSIRIHERTH
TGEKPYECKQCGKAFSYLNSFRTHEMIH
TGEKPFECKRCGKAFRSSSSFRLHERTH
TGQKPYHCKECGKAYSCRASFQRHMLTH
For completeness, there is a degenerate zinc finger in ZNF136 with the sequence “YGEKPDTRNQCWKPFSSHHSFRTHEIIH”
Why are the Zinc Fingers so different in sequence (except for the conserved amino acids)? To target a section of DNA, the zinc finger must be tuned to target it. Think of the zinc finger like a lock and DNA as a key that fits into the lock. In fact, for both the study of biology and medical applications, humans have a desire to make their own zinc fingers — like lock smiths. To bind a large segments of DNA, side-by-side zinc-fingers have to be tuned to their respective side-by-side DNA partners such as illustrated here:
By the way, there is a website that helps researchers construct the right amino acid sequence to make a zinc finger for a particular DNA target:
https://www.scripps.edu/barbas/zfdesign/zfdesignhome.php
Now, to visualize the critical/conserved amino acids, see the protein sequence here with highlights on “C” and “H” amino acids.
http://theskepticalzone.com/wp/wp-content/uploads/2019/04/znf136_zfC2H21.png
{kind=link}
Note one of the lines is not exactly like the other lines in that it is missing a “C”. This is the degenerate zinc finger mentioned above. So there is 1 degenerate zinc finger and 13 functional ones.
From this diagram it is apparent that the regular appearance of “C” and “H” is a violation of the law of large numbers, hence this pattern is not due to random point mutation alone. To “solve” this problem, Darwinist explain the pattern through segment duplication followed by some point mutation and natural selection, but this is not mechanically feasible either!
To understand why, let the reader first ponder the alignment I made of the Zinc Fingers in the ZNF136 protein using MEGA 6.0/MUSCLE software:
http://theskepticalzone.com/wp/wp-content/uploads/2019/04/znf136_zfC2H2_muscle1.png
{kind=link}
Then let the reader, consider the distance matrix generated by MEGA 6.0 which measures the number of nucleotide and percent differences between the zinc fingers.
http://www.creationevolutionuniversity.org/public_blogs/reddit/znf136_distance_matrix.xls
(All of the above results are reproducible, so I leave it to interested parties wanting to confirm the results to do so.)
For the duplication to work, at a bare minimum the right 84 nucleotide segment must be chosen, and then perfectly positioned for insertion so as not to break a pre-existing zinc finger. But supposing the duplication succeeds, why are the zinc finger’s conserved features involving “C” and “H” and other amino acids preserved and not eventually erased by point mutation given we obviously see the zinc fingers are different from each other. To preserve the “C” and “H” and other necessary amino acids in a zinc finger, the new zinc finger needs to be under selection. But in that case one is simply concocting a “just so” story for those newly minted zinc fingers without any respect for the difficulty of such a “just so” story being probable — and it is not probable!
To understand the problem of such a “just so” story, recall zinc fingers bind to DNA regions. Btw, this includes DNA regions such as ERVs! And ERVs are indicated to participate in the Stem Cell Pluripotency regulatory network:
https://www.nature.com/articles/nature13804
Further, the KRAB-ZnF protein is part of an incredibly complex machine that does chromatin modification (as shown above) by often attaching to ERV targets. But this would require that ERVs (or whatever DNA target) needs to co-evolve with the KRAB-ZnFs that attach to them!
Do these researchers even consider the fact such evolution would have to be instantaneous otherwise it would degrade function and not let the genome have the opportunity to adapt to the new accidental copy of the zinc finger because the duplication would immediately be selected against!
The above KRAB-ZnF complex is like a read/write head acting on Chromatin. Chromatin itself is an amazing mind-boggling design akin to computer ROM and RAM in one.
Again, the difficulty of evolution via random insertion/duplication mutations followed by point mutations is that such events would disrupt the binding of an already operational set of zinc fingers. For example, suppose we have an array of 10 zinc fingers side-by-side that collectively bind to a target DNA. Suppose one zinc-finger is duplicated and the number of zinc fingers is increased from 10 to 11. Oh well, the binding ability is broken or at best compromised, much like adding a single letter to a pre-existing password!
In sum, there is a violation of the law of large numbers in KRAB-ZnF proteins which is not explained by random mutation, nor random segment duplication followed by some point mutation and fixed by natural selection. Some other mechanism for the emergence of such proteins is indicated and would likely be indistinguishable from a miracle. Given the importance of such zinc finger proteins in the control of ERVs which are important in the stem cell pluripotency regulatory circuits, the origin of KRAB-ZnFs is even more miraculous.
I am curious Bill, do you honestly think that there is an equivalency to the way in which Sal and the rest of us are “talking over each other”, or do you know that phrasing it that way is a handy rhetorical gambit to make the two sides appear equivalent, in a “Trump on Charlottesville” kinda way?
Here’s Sal’s argument:
Ack! My brain hurts.
colewd
I suggest you read the OP, and the links therein. Especially the bits about creating DNA binding domains to order.
From the Opening Post
Is that the best you got? LOL! Just make crap up even when I said otherwise from the very start. Thanks for the entertainment.
Bill, thanks for showing. It’s not worth paying attention to their drivel. That’s example right there.
This is a teachable moment to see how the moderator of TSZ makes up crap:
but in contrast I said from the very opening the degnerate finger wasn’t functional.
Will he make a retraction now that’s been called out on attributing to me a claim I didn’t make?
Bill, don’t be bamboozled by this sort of crap. I may not take the time to call out all of it since it’s going to take a lot of time.
Don’t worry about defending me. I’m not here to advertise my work, I’m here to get editorial review and find out what sort of falsehoods, misrepresentations, and other crap my critics might throw at it so I can better compose my essays.
What I wrote.
My apologies, Sal. You did in fact notice that repeat 1 is not a full Zinc finger.
Now that we have that out of the way, perhaps you will ease up on the misrepresentation and address the remainder of everyone’s demonstrations of your errors.
Or not. Your choice.
A teachable moment:
This is how to criticize an argument when you can’t refute the actual points. Criticize something that wasn’t ever claimed or said. Here is an example:
Ah but DNA_jock seems to have the audacity to say stuff like that given I said from the beginning:
To say:
Means the degenerate one is NOT functional.
But this doesn’t stop DNA_Jock from saying:
Will he make a retraction? Any bets, or will he perpetuate the falsehood, pretend he wasn’t called out on it.
The other question is whether it’s worth my time if this is the best he’s got because he’s certainly not refuted the other problems I pointed out.
As far as the supposed clustering, the unrooted tree does a better job of identifying clusters, I didn’t have to notice or not notice, I let the computation made by evolutionary biologists do the talking as in:
Only if you root the tree like Evograd did.
The unrooted tree which isn’t as prejudiced, and the one I did, gave a different clustering.
There was a typo above where I used the #13 twice, namely
#13 499-526,
#13 471-442
when it should have been
#13 471-498
#14 499-526
But other than that, both graphs I generated refute DNA_Jock’s claim, whether #13 (above) or #14(corrected), the clustering isn’t as DNA_Jock says UNLESS one forces a rooting like evograd did which is imposing something of a pre-conceived phylogeny to begin with, and in the absence of other data,
that is circular reasoning.
The Neighbor Joining does a different clustering:
But if we drop neighbor joining, the actual distances from 4 are:
1 vs. 4 = 16
2 vs 4 = 12
3 vs 4 = 16
4 vs 4 = 0
5 vs 4 = 16
6 vs 4 = 13
7 vs 4 = 11
8 vs 4 = 15
9 vs 4 = 5
10 vs 4 = 15
11 vs 4 = 11
12 vs 4 = 12
13 vs 4 = 11
Moral: nearness is in the eye of the beholder and IT’S FREAKING IRRELEVANT TO THE PROBLEMS THAT I’VE POINTED OUT.
One can assume one phylogeny exists, it doesn’t refute the point that this is an improbable structure for the reasons stated.
Corrected graph:
Are you going to retract you howler that adding a zinc finger copy increases binding affinity for a PRE-EXISTING cis-regulatory element? 🙂
Do explain.
Really. This will be fun.
I’m all ears.
DNA_Jock claims making tandem IDENTICAL repeats increases binding affinity.
Well I was speaking of binding affinity to a PRE-EXISTING DNA target RELATIVE to the affinity the tandem repeat mutant will bind to.
I wasn’t speaking about some OTHER concoction where it might actually increase binding affinity. I was talking zinc-finger-to-DNA binding, as in a zinc-finger to DNA cis-regulatory element of a gene.
From:
http://zifit.partners.org/ZiFiT_v3.3/Program_use.aspx#_ZiFiT_ZF_Array
So the finger binding affinities are:
ZF3 = GCG
ZF2 = TGG
ZF1 = GCG
That’s a gross simplification because actually each ZF binds up to 4 bases and must over lap in binding with the adjacent ZF as mentioned above! But I’ll go with the simple diagram for now:
But lets suppose we had tandem repeat zinc fingers binding to the same motif : GCG
ZF3 = GCG
ZF2 = GCG
ZF1 = GCG
So the composite target of identical tandem repeats is:
GCGGCGGCG
not
GCGTGGGCG
Now suppose the original array was only 1 zinc finger that bound to GCG. Well that’s awfully non-specific and would have binding affinity to lots of places on the genome by random chance! But now lets add 2 tandem repeats (using the simplified logic which isn’t exactly correct because of the 4bp and overlap problem mentioned earlier by me). The binding target is now:
GCGGCGCGC
thus when the protein having only 1 zinc finger might previously have some chance of binding to
TTTGCGAAA
it will NOT increase the likelihood that the array of 3 tandem zinc fingers will bind to that relative to this target:
GCGGCGCGC
One might argue that it will could still bind to the old target of:
TTTGCGAAA
But if we include the problem of overlap mentioned above where non-symetric 4base DNA sequences are targets of the zinc finger, then increased binding is dubious.
What do I mean? The actual situation is the ZF can target 4 bases like:
ACGA
So a tandem 2 repeat zinc finger could bind this:
ACGACGA
where
ZF1 binds ACGA
ZF2 binds ACGA
ACGA has some symmetry between the A at the start and A at the end. But consider a non symmetric target for the original zinc finger:
ACGT
So a tandem repeat will have the following:
ZF1: ACGT
ZF2: ACGT
The composite recognition sequence would be:
ACG?ACG
where question mark is for how the 2 identical repeat tandem will handle the overlapping position.
Since DNA_jock apologized for falsely saying I failed to point out the degenerate zinc finger is non-functional, I will extend a little bit of charity.
This stuff is confusing, and it is possible DNA_Jock mis-understood what I said. I will attempt to clarify. If a solitary zinc finger targets this motif:
ACGA
then a partial list of DNA segments the zinc finger could park in the vicinity of could be (with the ZF binding target in caps, and the flanking DNA in lower case):
1 ACGA aaaa
2 ACGA aaac
3 ACGA aaag
4 ACGA aaat
5 ACGA aaca
6 ACGA aacc
7 ACGA aacg
8 ACGA aact
9 ACGA aaga
10 ACGA aagc
11 ACGA aagg
12 ACGA aagt
13 ACGA aata
14 ACGA aatc
15 ACGA aatg
16 ACGA aatt
16 ACGA aatt
17 ACGA acaa
18 ACGA acac
19 ACGA acag
20 ACGA acat
21 ACGA acca
22 ACGA accc
23 ACGA accg
24 ACGA acct
25 ACGA acga
……
64 ACGA tttt
Ok so now let’s say we have that tandem repeat described above what will the affinity be for?
ACGACGA
Well ACGACGA is #25 on the list above
So this sequence with have a binding affinity greater than its sisters in the above list. Hence the previous 63 other binding targets have reduced likelihood of being bound RELATIVE to sequnce
#25 ACGACGA
in that sense the other 63 sequences have a lessened affinity relative to the sequence that has the strongest affinity to the new tandem repeat.
That hopefully clarifies what I meant by reduced affinity. Since that verbage on my part may have caused confusion, I’ll think on how to phrase it differently.
But the fundamental point remains that a former potential function is now altered and even in this case, assuming HYPOTHETICALLY it was functional to target ACGA with one zinc finger, now 63 prior DNA sites have been rendered functionless by a “random tandem” repeat of a pre-existing zinc finger.
That’s the BAD JUJU I was talking about.
Inappropriately, I’m reminded of the Charge of the Light Brigade – both the poem and the event.
If you had a duplication of a protein segment with affinity for a particular DNA sequence, I really don’t get how you’d get reduced binding as a result. Some of the protein molecules would bind using site A, some site B. The two sites aren’t competing with each other within any given molecule. Displacement of a B-bound molecule by an A would be no different from an A shoving another A out the way. Detachment of a single molecule at B and re-attachment at A is still bound.
When bound by site A, there will be some interactions of B with flanking regions. And vice versa. This would often tend to increase the overall binding energy … wouldn’t It?
Yep. Imagine a piece of tape that you stick to something. One of the flanking sides of the tape has a bit of hair on it so it doesn’t stick as well as the rest, but it still sticks.
I was speaking in terms of RELATIVE binding affinities rather than absolute affinities.
In my preceding comment the binding affinity in my pedagogical/hypothetical model for the 64 sites was the same.
The affinity is 1/KD where KD is the dissociation constant.
Prior to the tandem repeat event
1/KD_1 = 1/KD_2 = …. 1/KD_25 .. 1/KD_64
after the tandem repeat event
1/KD_25 > 1/KD_1, 1/KD_2….1/KD_24, 1/KD26…1/KD_64
That’s what’s important. If this were not substantial enough of a difference, then we might expect the multi-zinc finger arrays to have their individual zinc fingers bind all over the place, but that apparently doesn’t happen — certainly the ZFIT consortium, when they intelligently design a zinc finger array for their custormers, they expect it to bind to the specified target, not have the individual zinc fingers randomly attach all over a genome!
But you don’t know that, you’re just asserting it would be rendered non functional. There’s just no reason to think that.
Relative to what? The singleton’s affinity? It can easily go up. It would be perverse to deny this.
You can’t decompose it in that per-site way. Two molecules have an overall affinity. It’s that which determines how well they stick together. Obviously, the bits and pieces of the molecules contribute to that overall affinity, by the electrostatic zone of influence of each bit of zwitterion or whatever, but it’s nonsensical to try and separate out the contribution of each DNA base.
If a single zinc finger targets a particular set of bases, why would a dimer go anywhere else?
The peculiar notion being peddled here is that, if a single finger targets a given sequence, a dimer will helplessly settle halfway between the two, in a perpetual – but weak! – arm-wrestle for the target. Before heading off and binding somewhere else instead, for which there is no affinity.
For convenience I will call 1/KD = KA
Do you think KA_25 > KA_1, KA_2, … ???
But let’s say a random tandem repeat event doesn’t make KA_25 THE chosen one, but rather KA_X
So in principle KA_X > KA_1, KA_2 ……KA_64
So even in that case my point is proven. Previously cis-regulatory targets are now not as likely to be bound as before. QED.
What the fuck are you talking about?
He’s saying that because he refuses, and or lacks the background/abilities, to even try and understand explanations to the contrary, he’s right.
Rumraket,
No, silly, he’s saying that if the protein binds somewhat better to a second site, there is less of it available to bind to the original cognate site.
We can discuss how funny that idea is after he has confirmed that this is his meaning.
I’ll admit that I was originally just hypothesizing from first principles re binding affinities, but it turns out…
stcordova,
Not sure you’re getting my point. If a single finger binds well to a particular sequence, why would the binding of a dimer be less strong? I mean, there may be steric interference or electrostatic effects between the monomers diminishing the substrate interaction, but that aside, to the extent that subunit B is shoving A out the way, or vice versa, they are doing so because of their mutual attraction to the single substrate.
The dimer binding is stronger than the monomer. That was the case in #25 ACGACGA above, or equivalently the dissociation is less for #25.
For that reason, with the same amount of the transciption factor concentration, more of the transcription will bind to #25 and less to the other 63 cis regulatory regions in my pedagogical example.
stcordova,
What will happen given this condition?
IF the other 63 sites were important, and now they are getting serviced less or not at all, in general this is the result:
stcordova,
This can certainly be true with transcription factors. There is a range of acceptable function or transcription rates however.
See, Rumraket, I told you that that was his point.
Do you want to tell him, or shall I, just how much of the protein will be bound to sequences that are completely unrelated to any target sequences?
Kudos to colewd for his “I don’t think that will matter” response.
Yeah Rumraket. You tell him right Jock, you said so yourself:
DNA_Jock even bolded it like that. Yeah. MORE SELECTIVELY. As in tighter to the new target to the exclusion of the set of old targets.
Well done!!!!! HAHAHA!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
You fail again in your misrepresentation. Now you owe me a second apology.
No Sal, read my grade school demonstration:
More selectively means a bigger difference between best and worst.
The “exclusion of the set of old targets” has nothing to do with what I wrote in either case.
When I wrote “sequences that are completely unrelated to any target sequences”, I meant precisely that.
DNA binding proteins spend a lot for their time bound to completely unrelated sequences, and a decent amount of time unbound, so the amount of time they spend bound to NEAR-matches doesn’t really matter too much.
This is a pedagogical representation of the cis-regulatory region prior to the tandem repeat, with the concsensus binding of the Zinc finger in CAPS:
1 ACGA aaa
…
25 ACGA cga
……
64 ACGA ttt
after a tandem repeat, will the cis-regulatory regions experience the same amount of binding by the zinc finger protein as before the Zinc Fingers were repeated? Not likely.
to use DNA_Jock’s words:
to #25 more so than the other 63 after the supposed improbable “random tandem” repeat event.
The problem remains, as I pointed out. About the only criticism now is just mincing of words.
Oh Sal, I covered this in my cartoon “Grade School” example two days ago.
You’re grade school example wasn’t what I was talking about. You refuted an argument I didn’t make.
LOL!. So #25 will have the same binding affinity as #1, #2,….#64.
I pointed out:
KA_25 > KA_1, KA_2…..
agree or disagree. And if not KA_25, some KA_X. And the point remains.
stcordova,
The fallacy of the Texas sharp shooter fallacy argument second edition 🙂
Why would it be the same amount? TFs are themselves subject to regulation, up and down. If the concentration goes down, because more is bound, more can be made.
DNA_Jock is invited to stated for the readers whether believes after the tandem repeat
KA_25 = KA_1, KA_2, …..KA_64
Sounds great. Will you be putting your presentation on t’youTube?
DNA_Jock is invited to stated for the readers whether believes after the tandem repeat
KA_25 > KA_1, KA_2, …..KA_64
and if not KA_25, maybe some KA_X
Surely DNA_Jock is not asserting after the tandem repeat of the zinc finger protein the relative ranking of the KA_S for each of the cis-regulatory regions will be the same after the tandem repeat?
And in Jock’s silly grade school example, the teacher should be fired since he asserted Entropy’s silly notion a random tandem will generate 13 zinc fingers covering exactly 84 nucleotides each. Random tandem makes more RANDOM tandems, not orderly tandem repeats.
Great idea.
Since DNA_Jock and Entropy keep saying how right they are, are they still swearing that the collective effect of mutliple random tandem ectopic duplication events over time will take some random protein and make the orderly pattern in the ZNF136 protein here that I highlighted in the opening post?
http://theskepticalzone.com/wp/wp-content/uploads/2019/04/znf136_zfC2H21.png
Here you go entropy, how about you take 1 KRAB domain, slap a solitary zinc finger on it and provide odds that a few iterations of RANDOM ectopic duplication of RANDOM length and RANDOM starting coordinates will make something orderly like:
http://theskepticalzone.com/wp/wp-content/uploads/2019/04/znf136_zfC2H21.png
Apparently neither you nor DNA_Jock have even given a guess at the odds.
I could throw a ZFP with 42 zinc fingers, but that might be really mean. 🙂
Oh Sal, we have covered your “The unreasonable precision of RANDOM TANDEM duplications” previously:
and
Actually, I am asserting that the relative ranking will have changed little, if we are talking about metazoans…
And your “KA_25 is now the best, nyah nyah nyah” “argument” was entirely anticipated by my “look, one of the other sites is now the best binder” exposition in my “grade school” example.
Please pay attention.
Thanks you for stating you position clearly. 🙂
Now that you’ve laid your cards down, let the reader appreciate some real science:
https://www.pnas.org/content/pnas/109/26/E1724.full.pdf
Well, this is awkward.
I encourage you to read the paper you referenced, Sal, in particular the bit about “search mode”.
For anyone-who-isn’t-Sal: “Search mode” corresponds to the Egr-1 protein bound to “sequences that are completely unrelated to any target sequences”, like I just explained.
For Sal, did you understand ANYTHING they wrote about “non-specific complexes”?
So let me describe the fine points as best we understand in the present literature.
So, what does that mean? Well the zinc finger array will prefer one sequence of DNA over another. It doesn’t mean it won’t bind to a non-specific sequence, but it won’t remain there as long as it will to the target sequence. This is dissociation (KD) and translocation.
And once the zinc finger array gets to the target sequence, it gives the complex a higher probability of forming and doing it’s business.
That’s my reading of this:
http://www.jbc.org/content/288/15/10616.full
The point is, whatever random tandem duplication events followed by point mutation happen, the newly evolved protein will preferentially target a different DNA sequence. This preferential targeting can’t happen at random, otherwise it is bad juju.
One can’t assume, naively as Entropy does, that random tandem ectopic duplications will naturally make zinc finger arrays easily and/or the results will be preferentially selected to go to fixation or evolve further. It is naive as expecting the re-targeting of pre-existing transcription factors to new cis-regulatory sites will necessarily be beneficial. BAD JUJU!
Or that rare set of random tandem ectopic duplications that are fortuitiously beneficial will emerge frequently enough to will overcome the waiting time problem that several evolutionary biologists (not just a creationist like Dr. Sanford) acknowledge is a real problem.
This point is normally where people would say “and therefore my solution to this problem is that…”
Oh I guess you forgot to mention the bind transiently and the TRANSLOCATE to other sequences to actually get to the target sequence in a sea of billions of non-specific sites.
More like obfuscated and distorted. Saving face now, DNA Jock?
You want to tell the readers with a straight face you expect random tandems to necessarily be functional changes? Do you want to suggest what proportion of such random events will result in functional improvements AND be selectively favorable?
You want to say with a straight face that binding to non-specific DNA sequences precludes the zinc finger array from eventually parking on the intended target. What the paper I provides shows is that the zinc finger arrays are like people searching for houses to live int. They go into a house briefly and leave if they don’t find what they are looking for. The visit and leave scenario is like zinc finger arrays binding to completely unrelated sequences. But when the target house is found, SHAZAM, the transcription factor finds its home!
Therefore my solution to this problem is God did it!