
Evolution of KRAB Zinc Finger Proteins vs. the Law of Large Numbers
There are patterns in biology that violate the law of large numbers, and thus suggests Intelligent Design or at the very least statistical miracles. The pattern involves KRAB-ZnF proteins that have multiple zinc finger domains side by side that are inexact copies of each other and would require a scenario of co-evolution of their DNA binding partners with every additional zinc-finger insertion — a scenario indistinguishable from a miracle.
The role of a zinc finger can be something like a clamp or a lock. Each zinc finger in a KRAB-ZnF protein is fine-tuned to connect with a DNA much like a lock (the zinc finger) can receive a key (like DNA). Here is a depiction of a KRAB-ZnF protein with 4 zinc fingers as part of a large chromatin modifying complex. The four zinc fingers are marked “ZN” and connect to DNA:
The evolutionary explanation of side-by-side repeated patterns of zinc fingers such as in KRAB-ZnF proteins shows a severe lack of critical thinking by evolutionary biologists who pretend “phylogenetic methods” are adequate explanations of mechanical feasibility of common descent.
To illustrate the problem, consider the KRAB-ZnF protein known as ZNF136. For reference, this is the amino acid sequence of ZNF136:
https://www.uniprot.org/uniprot/P52737.fasta
This is an amino acid fragment contained within the ZNF136 protein
TGEKLYDCKECGKTFFSLKRIRRHIITH
This short sequence is called a zinc finger domain which in 3D looks like this:
https://sciencescienceeverywhere.files.wordpress.com/2015/08/figure-11.jpg
{kind=link}
Wiki gives a description of the function of zinc fingers in proteins that have them:
http://wikipedia.org/wiki/Zinc_finger
A zinc finger domain requires two “C” amino acids and two “H” amino acids placed in the right positions. It requires a few other things too…
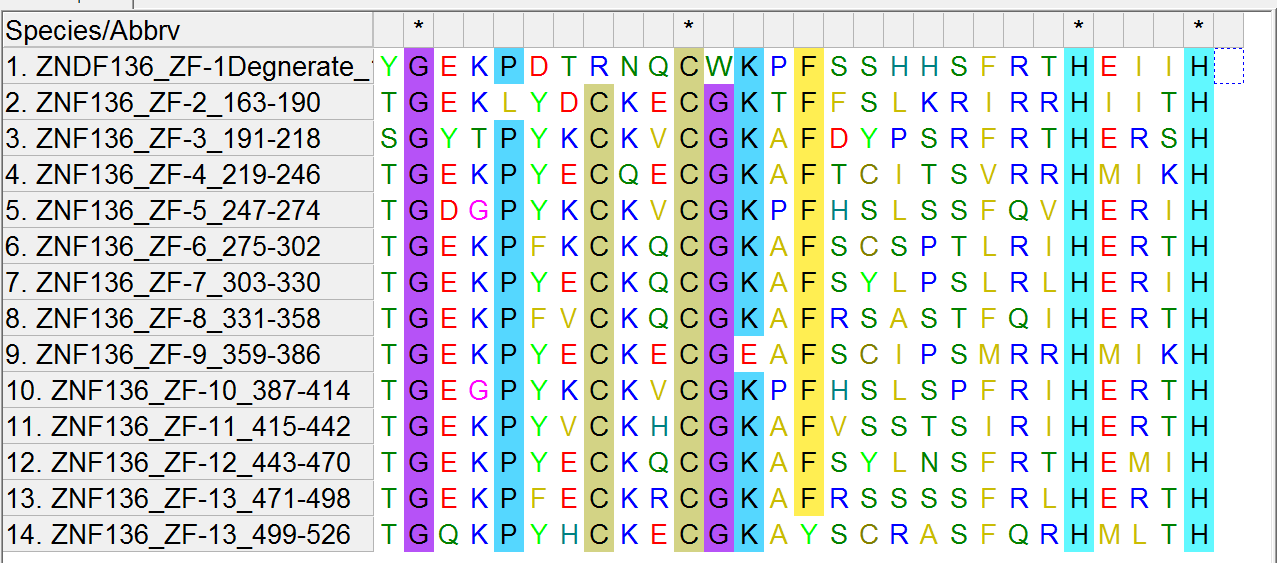
There are 13 zinc fingers in the ZNF136 protein and these are their sequences:
TGEKLYDCKECGKTFFSLKRIRRHIITH
SGYTPYKCKVCGKAFDYPSRFRTHERSH
TGEKPYECQECGKAFTCITSVRRHMIKH
TGDGPYKCKVCGKPFHSLSSFQVHERIH
TGEKPFKCKQCGKAFSCSPTLRIHERTH
TGEKPYECKQCGKAFSYLPSLRLHERIH
TGEKPFVCKQCGKAFRSASTFQIHERTH
TGEKPYECKECGEAFSCIPSMRRHMIKH
TGEGPYKCKVCGKPFHSLSPFRIHERTH
TGEKPYVCKHCGKAFVSSTSIRIHERTH
TGEKPYECKQCGKAFSYLNSFRTHEMIH
TGEKPFECKRCGKAFRSSSSFRLHERTH
TGQKPYHCKECGKAYSCRASFQRHMLTH
For completeness, there is a degenerate zinc finger in ZNF136 with the sequence “YGEKPDTRNQCWKPFSSHHSFRTHEIIH”
Why are the Zinc Fingers so different in sequence (except for the conserved amino acids)? To target a section of DNA, the zinc finger must be tuned to target it. Think of the zinc finger like a lock and DNA as a key that fits into the lock. In fact, for both the study of biology and medical applications, humans have a desire to make their own zinc fingers — like lock smiths. To bind a large segments of DNA, side-by-side zinc-fingers have to be tuned to their respective side-by-side DNA partners such as illustrated here:
By the way, there is a website that helps researchers construct the right amino acid sequence to make a zinc finger for a particular DNA target:
https://www.scripps.edu/barbas/zfdesign/zfdesignhome.php
Now, to visualize the critical/conserved amino acids, see the protein sequence here with highlights on “C” and “H” amino acids.
http://theskepticalzone.com/wp/wp-content/uploads/2019/04/znf136_zfC2H21.png
{kind=link}
Note one of the lines is not exactly like the other lines in that it is missing a “C”. This is the degenerate zinc finger mentioned above. So there is 1 degenerate zinc finger and 13 functional ones.
From this diagram it is apparent that the regular appearance of “C” and “H” is a violation of the law of large numbers, hence this pattern is not due to random point mutation alone. To “solve” this problem, Darwinist explain the pattern through segment duplication followed by some point mutation and natural selection, but this is not mechanically feasible either!
To understand why, let the reader first ponder the alignment I made of the Zinc Fingers in the ZNF136 protein using MEGA 6.0/MUSCLE software:
http://theskepticalzone.com/wp/wp-content/uploads/2019/04/znf136_zfC2H2_muscle1.png
{kind=link}
Then let the reader, consider the distance matrix generated by MEGA 6.0 which measures the number of nucleotide and percent differences between the zinc fingers.
http://www.creationevolutionuniversity.org/public_blogs/reddit/znf136_distance_matrix.xls
(All of the above results are reproducible, so I leave it to interested parties wanting to confirm the results to do so.)
For the duplication to work, at a bare minimum the right 84 nucleotide segment must be chosen, and then perfectly positioned for insertion so as not to break a pre-existing zinc finger. But supposing the duplication succeeds, why are the zinc finger’s conserved features involving “C” and “H” and other amino acids preserved and not eventually erased by point mutation given we obviously see the zinc fingers are different from each other. To preserve the “C” and “H” and other necessary amino acids in a zinc finger, the new zinc finger needs to be under selection. But in that case one is simply concocting a “just so” story for those newly minted zinc fingers without any respect for the difficulty of such a “just so” story being probable — and it is not probable!
To understand the problem of such a “just so” story, recall zinc fingers bind to DNA regions. Btw, this includes DNA regions such as ERVs! And ERVs are indicated to participate in the Stem Cell Pluripotency regulatory network:
https://www.nature.com/articles/nature13804
Further, the KRAB-ZnF protein is part of an incredibly complex machine that does chromatin modification (as shown above) by often attaching to ERV targets. But this would require that ERVs (or whatever DNA target) needs to co-evolve with the KRAB-ZnFs that attach to them!
Do these researchers even consider the fact such evolution would have to be instantaneous otherwise it would degrade function and not let the genome have the opportunity to adapt to the new accidental copy of the zinc finger because the duplication would immediately be selected against!
The above KRAB-ZnF complex is like a read/write head acting on Chromatin. Chromatin itself is an amazing mind-boggling design akin to computer ROM and RAM in one.
Again, the difficulty of evolution via random insertion/duplication mutations followed by point mutations is that such events would disrupt the binding of an already operational set of zinc fingers. For example, suppose we have an array of 10 zinc fingers side-by-side that collectively bind to a target DNA. Suppose one zinc-finger is duplicated and the number of zinc fingers is increased from 10 to 11. Oh well, the binding ability is broken or at best compromised, much like adding a single letter to a pre-existing password!
In sum, there is a violation of the law of large numbers in KRAB-ZnF proteins which is not explained by random mutation, nor random segment duplication followed by some point mutation and fixed by natural selection. Some other mechanism for the emergence of such proteins is indicated and would likely be indistinguishable from a miracle. Given the importance of such zinc finger proteins in the control of ERVs which are important in the stem cell pluripotency regulatory circuits, the origin of KRAB-ZnFs is even more miraculous.
Actually it doesn’t have to be always damaging. Simple drift will drift most changes out, even slightly “beneficial” ones. This is the problem of Gambler’s ruin.
The probability of fixation for deleterious to slightly advantageous mutations is on the order of 1/Ne, where Ne is effective population size. These are some of the inputs needed to make a waiting time calculation for a single coordinated system. Now, one might rightly argue there are more than one opportunities for other coordinated systems — as in more slot machines to be played. But this is counter balanced if there are a lot of coordinated changes required when evolving one creature to another.
I reviewed my numbers with John Sanford’s waiting time model yesterday, and he said the numbers for number of nucleotide changes in his paper were very conservative, smaller than those indicated by the zinc finger arrays I’m considering. So the waiting time problem would be very much more substantial for human-ape-like populations.
Dr. Sanford’s waiting time figure was 85 million years. Durrett and Schmidt (no friends of ID) put theirs on the order of 200 million years. Behe and Snoke was about 10^15 years.
Because Zinc Finger bind probabilities are better studied than Doug Axe’s beta lactamase studies, I’m suggesting Zinc fingers binding is a better model for estimating probabilities of specific binding interactions. And hence I’m trying to communicate this to the ID community as a model for a certain class of waiting time calculations.
With respect specifically to KRAB-ZnF proteins that repress transposon expression — there are some considerations.
First, we don’t know how bad the effect is if the transposons aren’t repressed. Would this be a lethal situation and hence not opportunity to evolve KRAB-ZnF proteins to suppress the transposons?
Next, a single zinc finger can specify only about 3 DNA bases. It is undesirable for a DNA binding protein to use 1 zinc finger to bind to DNA since it can bind to 1 out 64 (4^3). That would be a sizeable number of locations on a genome as big as a human genome . Even 2 repeat array would bind to 1 out of 4096 (4^6) which would still on the order of 800,000 locations on the human genome — which is more than the number of genes.
A 3 repeat array seems to be a good starting number for a Transcription factor. By way of comparison, TFiiiA has 10 zinc fingers and can bind to 55 bases!
One minor point is that the binding affinity is very much stronger in some experiments going from 1 to 6 repeats — on the order of a 6,000 fold change. Suggesting also a solitary zinc finger might not attach well anyway unless concentrations of the protein are changed appropriately.
My point, even starting with a solitary zinc finger is a dubious proposition for suggesting evolving function from a simpler gene.
The numbers just don’t seem feasible to me.
Agreed. It isn’t about you, your qualifications, motivations or character. What the thread is about is some claims made by yourself. Unfortunately, these claims, being made by yourself, cannot but reflect your lack of qualifications, understanding, education, background and motivation. Me and others have tried to help you understand the problems that have thus permeated into those claims. Unfortunately, yet again, the explanations, no matter how detailed, haven’t reached you because of your lack of qualifications, understanding, education, background and motivation. So, agreed again, the thread is not about all of that. However, all of that has an effect on the discussion. It’s unavoidable.
See Salvador? Others got it:
Seems like the problem is you, your character, background, education, motivation …
It doesn’t work Salvador, because there’s plenty of explanations from me to you in this very thread. Yet, you said yourself that you don’t read what I write:
Thus, I’m not attacking you. You confirmed what was already evident from your self-ridiculing “answers,” that the problem is that you don’t read, and the little you do, you don’t give it much thought. Yet you were pretending that you read it all, only to offer “answers” showing abysmal lack of understanding.
You said it yourself, so stop playing the victim. It shows lack of character.
Sal gives us another demonstration of selective reading with no consideration of context:
Let’s see that paragraph, shall we?
Yep. That’s evidence for a lot of neutral, semi-neutral, and perhaps even beneficial, juju.
It’s that word ‘most’. You treat it as a synonym for ‘all’ again. Most mutations aren’t beneficial, most that are drift out. Not in dispute, but evolution does not proceed by those.
I dare say, but they aren’t a problem for most evolutionary biologists – those who came up with the very equations and models you and Sanford utilise. Why do you think that might be?
And some of them believe in God.
Hi,
I see the similarity of Chimps and Humans and apes in when I’m browsing genes. Also the morphology is extremely close. Michael Denton, who is a quasi ID proponents, said when he was in medical school, he was dissecting human and ape cadavers and that turned him into an atheist because the similarity was so powerful. It would be years later he wrote Evolution a Theory in Crisis as he examined the facts more and came to a middle ground between unguided common descent and some sort of anthropic kind of Intelligent Design.
As I said, I used to be an evolutionist, and there is an obvious set of transitional steps from bacteria to humans even from extant organisms. I’m not saying the leaps don’t require miracles, but the transitions are there.
The waiting time problem has been put forward as evidence against Chimp/Human common ancestry. It’s a very esoteric argument.
In contrast there are sort of brute force kind of transitions like from unicellular creatures to an animal, or prokaryote/eukaryote transitions.
The point is, if one can accept special creation, such as animals, then one can accept special creation for other things if the improbability is strong enough. I thought the ape-to-man transition is the weakest link in the creationist claim since the similarities are brutally strong. Humans are surely more similar to chimps than they are to starfish or trees!
I found a nice domain diagram of collagen which I will reference in my presentation.
It’s from the USDA of all places!
https://pubag.nal.usda.gov/catalog/285217
CLICK TO ENLARGE:
http://theskepticalzone.com/wp/wp-content/uploads/2019/04/collagen4_domains-1024×401.png
stcordova,
Who are you talking to at this point?
Do you have a list of what was created and what evolved?
stcordova,
This transition by reproduction and isolated populations has been sold as a fact by Moran, Dawkins etc.
How would you confirm this hypothesis?
Consilience of independent phylogenies.
“The ape to man transition is the weakest link in the Creationist claim”. A remarkable admission – it’s the only one most of them give a damn about.
Yup. Most creationist want to think they aren’t mere animals based on biology. But the Bible teaches
As you said:
That’s true, and it’s lamentable to me because humans are VERY similar to chimps. They want to argue against common descent and pick the strongest example that suggests common descent!
They could instead pick the difference between bacteria and animals, since that entails at least two major transitions:
1. prokaryote/eukaryote
2. unicellular creature to animal
Thanks. Most creationists won’t say that, and that’s not good for them to do so.
I see many genes that are 99 to 100% identical between humans and chimps, at least in the exons. The question of the number of orphans between them is still open. BUT, even assuming lots of orphans, we are very similar to chimps relative to starfish or butterflies or blades of grass.
The presentation was inspired by papers on Promiscuous Domains. By way of extension there could be argued promiscuous PTM motifs, miRNA motifs, transcription factor motifs, lnc/lincRNA chromatin modifying motifs, localization sequence motifs, enahnacer motifs (like vitamin D receptor).
Now the motifs are generally not exact sequences, but follow apparent architectural principles that enable some sort of connection (binding).
Sooo, how does this relate to proteins? Well proteins are coded from DNA/RNA , and the miRNAs, as far as I know target the translated as well as untranslated regions. Hence the protein sequence is under constraint to allow the DNA/RNA that codes it to be regulated by miRNAs.
So are miRNA binding motifs promiscuous among non-homologous genes? Remains to be seen!
https://en.wikipedia.org/wiki/MicroRNA
Here is an miRNA interaction network.
http://www.bloodjournal.org/content/bloodjournal/suppl/2010/03/01/blood-2009-08-237495.DC1/FigureS1.jpg?sso-checked=true
There may be promiscuous motifs in proteins that have post translational modifications. For example the a kinase is able to phosphorylate numerous different kinds of proteins. Are the phosphorylations functionless accidents or are they functional?
The targets of things like phosphorylation don’t entail exact sequences, but obey some sort of architectural principle. So this is a “promiscuous motif” that may appear on several phylogeneticaly unrelated proteins.
Here is an example of the proteins that are the target of a single kinase. CDK1 targets 75 proteins in yeast. Here is a sample of some of them:
https://media.springernature.com/original/springer-static/image/art%3A10.1186%2F1747-1028-5-11/MediaObjects/13008_2010_Article_106_Fig5_HTML.jpg
Then there are Chromatin modifiers and promiscuous motifs on DNA. The HOTAIR lnc/lincRNA and the Polycomb Repression complex binds to 832 locations on the DNA? I saw the supplemental tables describing the binding sites — spread over several chromosomes! How did that happen? These control things like the quality of skin on the eyelids vs. souls of the feet.
This was the experiment that did a mapping of the HOTAIR/PRC2 connection points.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3249421/#SD2
Below is the diagram showing the chromatin modifying machine is not trivial. It’s hard to believe the 832 targets are random accidents.
Here is a paper on a single transcription factor regulating mutliple phylogenetically remotely related genes. How did that happen? This is suggestive of a promiscuous motif:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2710871/
stcordova,
Your comment betrays some classic ‘evolution-blindness’. If a process of gradual divergence is in train, it is inevitable that recent separations will show less difference than more distant ones. In view of this, it would be mistaken to take the substantial differences between – say – starfish and dandelions as definitively indicative of a genetic discontinuity between them. Particularly when there remains substantial evidence of continuity.
If one buys the evidence of phylogeny at the levels of genus or family, one would expect this to be a tool one could use to investigate the point at which discontinuity arises at broader taxonomic ranks. But it doesn’t find it. Small discontinuities are easily visible. Instances of HGT, of mutation, insertion, deletion, transposition and inversion pop up against the genetic background, and are informative of the placement of nodes. The tool appears capable of the necessary resolution.
So at some point – a point Creationists seem unable to locate – there should be a discontinuity. Rather than shading into greater discrepancy as one widens the taxonomic net, one should reach bedrock. The ability to detect tree-like patterns in data, uncontroversial at low levels, continues throughout. Therefore rather than an ad hoc explanation, in which genetic relationship gives way to Design choice at some undetectable point for some Biblical rationale, it seems reasonable, absent other data, to assume genetic relationship throughout.
Colewd admits that speculating or even having an opinion regarding when the designer acted is “above his pay grade” but they have no problem insisting it did act.
Odd how they know it acted but cannot say when or how or to what end. Yet they know it happened.
Seems like the literal definition of faith to me. And yet they persist, as per this OP, in trying to make it into science.
Revised Title of my Presentation:
Polyconstraints, Promiscuous Protein Domains and DNA Motifs, Probability Pedagogy and Practice
I found a nice diagram of Promiscuous Protein Domains in the Bromo Proteins. Bromo was supposedly named after the god Brahma, it has nothing to do with the element Bromine….
At the top of the diagram are normal domains. At the bottom are cancer-associated domains that were shuffled and cobbled together — obvious bad juju if domains are translocated around randomly. It nicely shows the architecture of higher eukaryotic proteins that are poly functional. Some papers suggest this polyfunctionality in higher eukaryotic proteins solves the G-value paradox — (i.e. how can human have fewer genes than rice!). Anyways, enjoy:
CLICK TO ENLARGE:
http://theskepticalzone.com/wp/wp-content/uploads/2019/04/bromo_domain_proteins.jpg
Zinc fingers enable proteins to connect to DNA. There are connectors that enable proteins to connect to other proteins like the PDZ domain. I will show a few of these graphs to illustrate the promiscuity of the protein domains, with PDZ and the zinc fingers and the SH3 domains among those that are promiscuous.
CLICK TO ENLARGE:
http://theskepticalzone.com/wp/wp-content/uploads/2019/04/pdz_domains_wiki.jpg
These are some proteins with SH3 domains. One can see a lot of domain shuffling:
Oops, saw your response just now. Thanks for replying.
Here are slides from my 2 1.5-hour presentations:
http://www.creationevolutionuniversity.org/public_blogs/reddit/promiscuous_domains_part_1.pptx
http://www.creationevolutionuniversity.org/public_blogs/reddit/promiscuous_domains_part_2_r1.pptx
Thanks to all here who gave me editorial feedback and helping my presentation be a resounding success!