
Background
For the past month or so I’ve been investigating the claim that the phylogenetic signal is evidence that a dataset shares common descent.
Supposedly, the phylogenetic signal is one of, if not the, strongest pieces of evidence for common descent. It is one of the first of the 29+ evidences for evolution offered over at Talk Origins (TO).
Quoting from the article:
The degree to which a given phylogeny displays a unique, well-supported, objective nested hierarchy can be rigorously quantified. Several different statistical tests have been developed for determining whether a phylogeny has a subjective or objective nested hierarchy, or whether a given nested hierarchy could have been generated by a chance process instead of a genealogical process (Swofford 1996, p. 504). These tests measure the degree of “cladistic hierarchical structure” (also known as the “phylogenetic signal”) in a phylogeny, and phylogenies based upon true genealogical processes give high values of hierarchical structure, whereas subjective phylogenies that have only apparent hierarchical structure (like a phylogeny of cars, for example) give low values (Archie 1989; Faith and Cranston 1991; Farris 1989; Felsenstein 1985; Hillis 1991; Hillis and Huelsenbeck 1992; Huelsenbeck et al. 2001; Klassen et al. 1991).
http://www.talkorigins.org/faqs/comdesc/section1.html#nested_hierarchy
I’ve been skeptical of this claim. A tree is just one kind of directed acyclic graph (DAG), and my hunch is many kinds of DAGs will also score highly on metrics for phylogenetic signal. I picked one metric, the consistency index (CI), which according to Klassen 1991 is the most widely used metric. It also is the featured metric in the above TO article. Plus, it is very simple to calculate. So, I’ve focused my efforts on the CI metric.
Result
What I have found is that my hunch is correct. It is simple to create a DAG that scores highly in CI, well within the range of published CI scores for real datasets.
Consequently, it is incorrect to say the phylogenetic signal is strong evidence for evolution. In particular, this claim is provably false (as I have proven here):
Phylogenies based upon true genealogical processes give high values of hierarchical structure, whereas subjective phylogenies that have only apparent hierarchical structure give low values.
http://www.talkorigins.org/faqs/comdesc/section1.html#nested_hierarchy
How have I proven it false? I generate DNA sequences from directed acyclic graphs, and the trees derived from these sequences using well established methods produce CI scores well within published ranges. Here are two such experiments plotted on the chart from Klassen 1991:
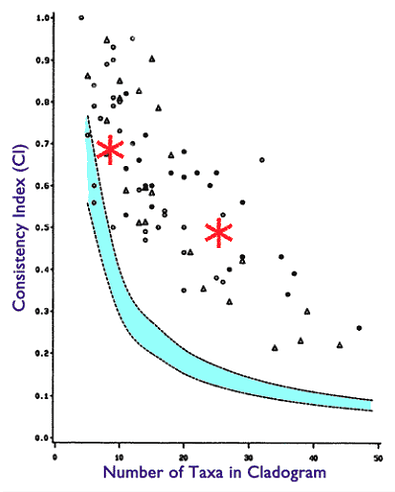
This is a phylogeny with very high value of hierarchical structure not generated from a true genealogical process.
Methods
To reproduce my results you can run the DAG dataset generator here: https://repl.it/@EricHolloway/Phylogenetic-Signal-Fallacy-Nucleotide-Level
Take the generated DNA sequences, which are in FASTA format, and paste them into the ClustalW online tool.
Take the results of the ClustalW tool, and use the PAUP software to generate trees and measure CI scores. You’ll need to fiddle with the NEX file format, so to save you the trouble, I’ve included an already created NEX file that I’ve generated from the aforementioned process, which you can pop into PAUP.
Once you load a NEX file into PAUP, here are the steps to generate trees, and then measure CI.
- Press “Generate Trees” in the “Trees” menu.
- Press the “OK” button.
- Press “Describe Trees” in the “Trees” menu.
- Press the “Describe” button.
- You will see something like the following:
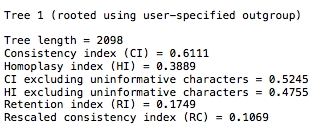
You will find the 27 taxa in the file will generate CI scores in the range of 0.48-0.53. If you look at the Klassen 1991 chart, you will see this is well within published scores for that number of taxa.
Conclusion
So, what is my takeaway from this?
Basically, highly statistically significant CI scores do not indicate common descent. They can just as easily be generated by a DAG. Therefore, we cannot infer common descent from high CI.
Furthermore, insofar as CI is representative of the state of phylogenetic signal measurement, my result undermines the more general claim that phylogenetic signal indicates common descent.
As such, the Talk Origin’s claim that the nested hierarchy of species is well attested by the data is highly questionable if not outright false, and should be retracted as evidence for evolution until such time as a much more rigorous analysis with DAG eliminating controls is established.
Addendum
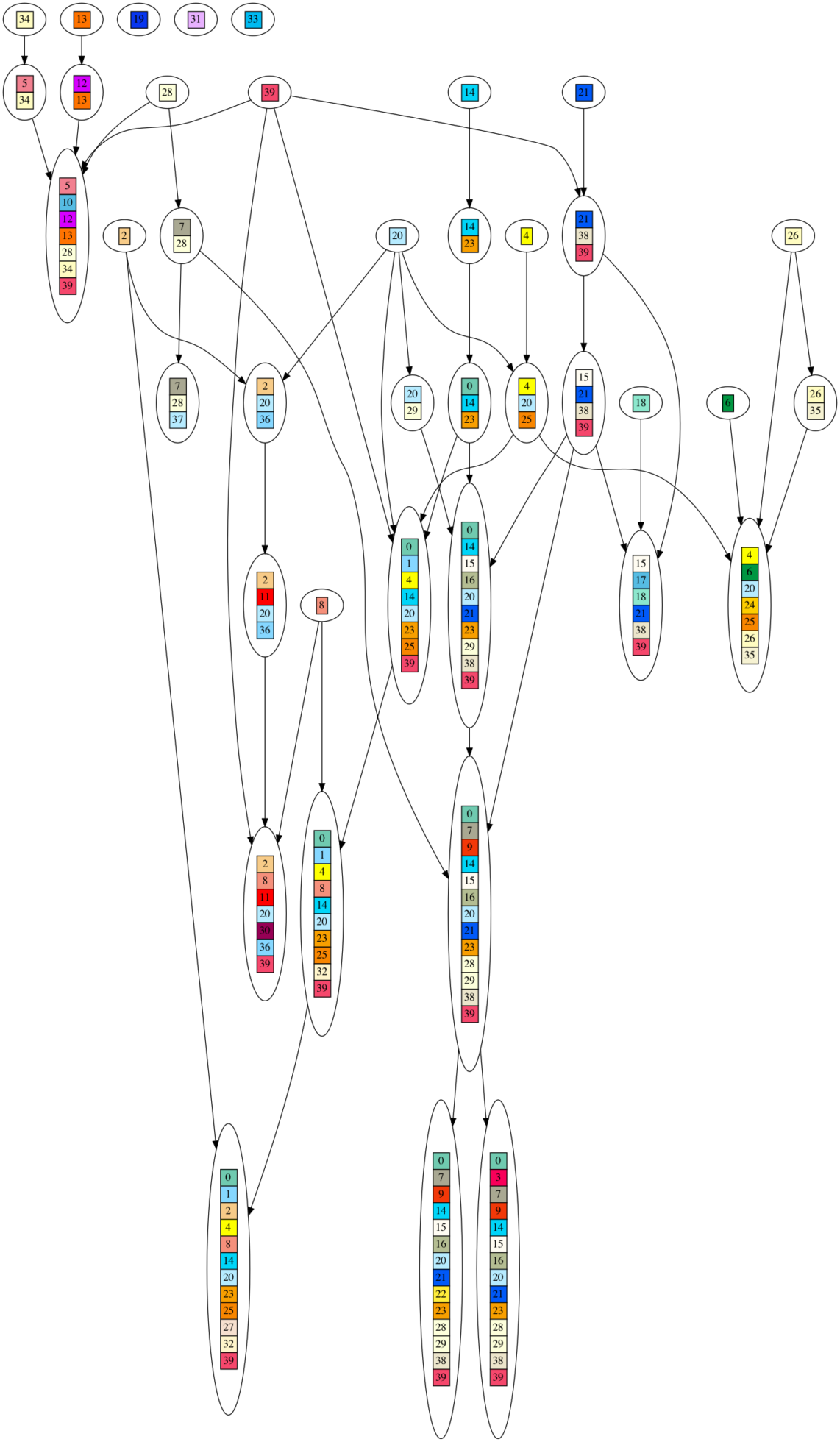
To visually illustrate what I mean by a DAG generating the DNA sequences, here is a graph of one such DAG. Each colored/numbered box represents a gene, which is replaced by a unique, randomly generated (uniform over ‘GATC’) DNA sequence of 20-30 letters long in post processing. Arrows indicate when ‘ancestor’ gene sets are combined into larger gene sets. If you look closely, you will see each gene set contains the union of all incoming gene sets, plus one new gene. As you can see, this looks nothing at all like an evolutionary process, yet it produces very high phylogenetic signal as measured by the consistency index (CI) metric.
John Harshman,
Here’s the link
Too bad nobody took me up on my bet. LOL
Holloway, do you think your god approves you blatant dishonesty?
Eric Holloway, employed to bullshit. LOL
Rumraket,
It’s not just a job. It’s his calling.
Alan Fox,
That’s some mighty strong gibberish. Is that where he posts the stuff he’s ashamed to post here, or what?
The site list of authors should give us a hint. I get the impression there’s an effort to present a more coherent and intellectual front for “Intelligent Design”*. Whether the effort is working is another question.
Why Eric posts here? It gives him an opportunity for feedback that echo chambers don’t provide without too much exposure if things go a bit belly-up.
ETA * than Evolution News & Views or Uncommon Descent.
I just had the brainwave that the reason I’m able to get these high scores with DAGs is because of what the article explains, all the CI scores are actually indicative of large amounts of homoplasy (1-CI = homoplasy index), which can most easily be described by a DAG. And since in the higher taxa counts the HI >> CI, this means a DAG will fit the data much better than a tree.
The key point is that a perfect tree scores a 1 on the CI metric, and almost none of the studies show perfect tree scores. If it is not a perfect tree, then it fits one of the non-tree DAGs I’ve been mentioning. If the CI is just a little less than 1, then the DAG is probably not too far from a perfect tree. But most of the studies with large numbers of taxa have a CI much, much less than 1, so the DAG is very different from a perfect tree, and is more analogous to the haphazard graph you’d get from human invention, or for similar reasons, the library dependency graph in computer code.
If you refer to the couple explanations I’ve posted, the colored boxes are ‘modules’ which can contain multiple characters (either 0/1 presence/absence of property, or GATC for a base letter). So, to avoid the whole alignment discussion, I went back to the binary situation and just used 0/1 and no gaps. The modules will contain multiple positions with 0 or 1, hence there are fewer modules than characters in the matrix.
It probably would be best if I wrote an article explaining exactly what is going on here.
We can call it a generator instead. There is some element of simulation going on, since there is an accumulation of items as one follows a path through the DAG, supposed to be analogous to how, say, human inventions will accumulate features.
Anyways, as someone else mentioned, the key point here is the structure, in particular that the generating structure is not a tree.
Eric, you are not answering Tom’s questions:
If you are simulating how human inventions accumulate features, why then apply it to sequence data?
That would be difficult, as you clearly don’t understand what is going on. You clearly have no idea what a presence/absence character is, what a 0 or 1 is supposed to indicate, or what, apparently, a taxon is. I’m sure, however, that you will continue to forge ahead in the supreme confidence that you know perfectly well what you’re doing. Your confidence seems impenetrable despite the clear history in this very thread of your multiple failures to do what you imagine you are doing.
Tom is right that it should not be called a simulator. Let’s call it a generator. Or a somethingnator.
The point is the graph structure.
Still relevant to sequence data. E.g. computer code is sequence data, and accumulates features, which show up as indel events and character substitutions in the git history.
I would say it probably doesn’t matter too much the level of detail of my understanding. The important thing is I can create a dataset from a non tree structure that scores highly on these metrics, hence showing we cannot infer a tree structure from high scores. That’s about all that I need to do here. Unless you can explain with specifics where you see a particular problem instead of vague obscurantism. At this point I believe I’ve explained what is happening in sufficient detail you can identify any problematic portion if you are actually able to find any issues. I’m doubtful you can, which is why you are just resorting to “you don’t know what you are doing” obscurantism.
That you think you’ve explained it is another indicator of how lost you are. Your last iteration was entirely undecipherable; looking at the alignment it was impossible to determine whether it actually was an alignment or reflected the process you claimed, which was itself not adequately explained.
The last comprehensible result you produced was due to bad alignment in which some bits were perfectly aligned (zero homoplasy) and some bits were poorly aligned (lots of homoplasy), the combination producing an intermediate value. But that told us nothing of value, only that mixing two sorts of data gives something in the middle.
EricMH,
Speaking as a strictly amateur sideline sniper, I’d say that if you have any common descent at all, you will generate something with more ‘treeness’ than something in which it is absent, because descent gives trees. ‘Perfect’ descent would give a ‘perfect’ tree, but partial situations with confounding factors – eg homoplasy or HGT – are also possible. So you need a model that excludes any descent processes.
And another week goes by. Apparently Eric’s only response will be to assert that it’s not important that he know what he’s doing.
He was probably eyeballing it.
On the bright side, he’ll probably publish in BioComplexity and you can respond there. 🙂
You could always write a program that declares Eric the winner. Programs are the gold standard, right?
Yup. Just ask Richard Dawkins. I could indeed write a “Weasel” program that declares Eric the winner.
You would probably declare “garbage in garbage out.” Or not.
What do you really think about programs which produce the intended outcome?
Designed?
That’s not what ‘Weasel’ does, as you well know.
All programs are designed, as far as I am aware.
All scientific models are designed, as far as I am aware.
Do you think that programs that are carefully designed to mimic the behavior of natural processes are somehow thereby misleading us?
I see that the troll is getting plenty of nourishment right now. Oddly, when fed, trolls only get hungrier.
Certainly not! So long as we agree natural processes are designed to produce certain outcomes.
Troll hungry.
I’m going to guess that your ‘job’ does not involve logic?
Arrogant much?
Anything further you want on my end? I offered an article explaining my method in more detail, but you said that is unnecessary. I don’t see anything further to do here.
Another ID-Creationist declares victory and runs for the door after getting his latest brain farting nonsense destroyed. What a surprise. 🙂
Depends on what this means. For example, if the program is intended to simulate random mutations and selection, the intended outcome could be: that the program really uses some randomizer to produce mutations and then selects and reproduces the best fitted things. In that case the program does what it should do. If the intended outcome is supposed to be: let’s see how far it can go, then the intended outcome would be whatever comes out, which might not necessarily be what we’d expect or like it to be.
Designed? The program/simulation is designed by definition. Their outcomes, the things we’re asking the simulation about, is what we’re after. Personally, I do not like simulations. Too easy to make fools out of ourselves. They have to be checked very carefully. However, sometimes there’s little more that can be done.
In Eric’s case here, the problems are built in from the very premises and presumed foundations, not just of the simulation, but of the strawman he’s built to then burn.
Can you explain the specific technical flaw you see in this particular work?
Yes. Genuine engagement with the problems in your “simulation” that have been pointed out here. An acknowledgment that it would indeed be useful, indeed important, for you to have a clear idea of what you were doing. An explanation of what your simulation is proposed to simulate.
A further description of your method would not be very useful, as the result clearly fails to do anything like what you’ve already claimed for it. Even your attempt at simple, binary presence/absence characters isn’t what you think it is.
See many previous comments here, none acknowledged in any substantive way.
And another week goes by. Just checking in.
I don’t have anything constructive at this point, but this post remains on my radar.
EricMH,
Why don’t you have anything constructive? Why have you never responded about the errors in your three previous attempts at a data generation program?
Sorry, just poking this thread to see if the stimulus produces a response. Has Eric abandoned his experiments?
John Harshman,
Feel free to poke away.
I read through the comments again, and as before I don’t see anything actionable. You state multiple times that the alignments are meaningless. Perhaps they are, but I’m not sure why. When I switch to binary datasets, you say I don’t know what I’m doing. Most assuredly so, but again that doesn’t give me much tangible to work on.
So, as I see it my only way forward is to read through the literature and datasets myself, and develop my own understanding of what a ‘real’ dataset looks like, and the delta between that and what my code generates. In which case, there is nothing left for me to do on this thread until such a day as I have such an understanding arrives.
My only conclusion from all of this is that understanding what a phylogenetic signal means is non trivial. The obvious interpretation that *only* datasets generated from tree shaped graphs generated phylogenetic signals is not straightforwardly true. There is a whole lot of extra expertise that seems to be required to make the phylogenetic signal analysis a reliable indicator of tree like graphs. Which makes it another piece of evidence for common descent that is out of the reach of amateur verification.
My bigger project is to find a very reliable piece of evidence for common descent that I can verify myself. So far every candidate evidence has turned out to be non trivial to support common descent, and require a lot of extra scientific expertise that I do not have. This is different than most other rigorous fields of science I am familiar with, where fundamental results often can be verified by complete amateurs. This is especially the case with my own field of expertise in computer science.
Do you have time for a basic course in biology? I’m sure that would help.
EricMH,
BTW Eric, are you sticking to your prediction of a Trump landslide?
They’re meaningless because homologous sites do not appear in the same columns of your alignments. In your first alignment, that’s because Clustal did a bad jjob. In your second alignment (where you introduce gaps into the actual sequences), homologous sites seem randomly scrambled.
Your binary characters do not correspond to single events, as it appears that multiple characters correspond to each claimed event. The nature of these multiple characters is opaque.
It’s unfortunate that this is all you get from our discussion, but I can only lead you to water; I can’t make you drink.
Here. Try something simple, involving crocodiles.
Another week, another bump. How you doin’, Eric?
Last try, I promise. Eric, you there?
Saw you latest response. I think I did not communicate clearly what is happening with the binary dataset. There is only a single character for each event, but in the interest of preserving space, long non-branching chains of events are lumped into a single box. That’s probably what is leading to the confusion.
I’ll look at your crocodile paper. Is it possible to get the dataset? Glancing through the paper I did not see an obvious link to the dataset.
@Alan Fox, is there a particular piece of evidence from a basic biology course that you think is really solid evidence for evolution? If so, I’ll look into it. I cannot of any good evidence from my high school GCSE biology class. For instance, we were fed the Haeckel’s embryo ontogeny recapitulates phylogeny, which is known to be a falsified dataset. We were also taught the moths experiment, which is also falsified evidence. We were taught evolution forms a tree, which it does not.
As for the Trump vote, be it on record I don’t like the guy, nor did I vote for him in any election, and don’t want him as my president. However, I do think this current election result is highly suspicious.
My personal opinion is Trump did win in a landslide, and vote fraud stole the election from him. Only one piece of concrete evidence I have is from a supposed timeseries of the Georgia vote, which showed a clear spike of ‘vote bundles’ that increased Biden’s advantage by 4800 votes over more than 10 times, which is easily enough to steal the state, since the margin is only around 10k votes.
Attached is a histogram of the ‘vote bundle’ deltas, divided by 1200. You can see a clear spike over the -4 tic on the x axis.
For comparison, here is the same histogram for Trump’s side, which shows no such spike.
Assuming the dataset I have is legitimate, and caveat is I have not been able to track it to an official source, it is apparently an anonymous leak, I deem this highly suspicious. It’s hard to explain a legitimate reason why deltas of vote bundles hover around 4800 for only one of the two candidates, and the spike seems too large in comparison to the rest of the data to just be random fluctuation. On the other hand, it’s easier to explain if it is a subtle manipulation of the vote that is attempting to avoid detection.
The best evidence is, for me, just looking at the world beyond our concrete cocoons. Do you have access to a garden, some growing space? Just clear a spot and watch what happens.
Given the decentralized state-run voting system, how was this organized? And why the superfluous 7 million Biden votes?
WhatThat has only aroused and fed suspicion. Your conspiracy theory demands collusion and conspiracy on an unprecedented scale.ETA
I found your data source, Eric, and there’s no need to get all fancy and math-y about this: at 11/4/20 06:12 UTC, Trump’s tally went from 2,275,266 to 2,264,513 !! They stole 10,753 votes from him !!
As the author notes: “Sorry, Democrats: this is what we call DNA-level statistical proof of fraud.”.
Strangely, none of these discrepancies were picked up in the hand audit of the entire state…
I mean, seriously, if you are going to promulgate a dishonest conspiracy theory, trying to undermine democracy, at least pick a state that was NOT subject to a hand audit, FFS! Also, “I couldn’t find the primary source, so I am just trusting some rando on the interwebs” is SO you, Eric.
Oh, I forgot to mention, at 11/4/20 06:12 UTC, Biden’s tally went down by 17,866.
That timestamped NY Times (yes, that was the source!) table is not displaying what you think it is displaying.
If you want a really DNA-level proof of fraud, how about the fact that your NYT data table has 51 non zero rows between 03:04 and 21:59 UTC on the 6th: they add 3,576 votes for Biden and 3,576 votes for Trump. Each of the 51 entries are IDENTICAL. Apply your ID-math to that result!
Eric,
Try modeling a process in which someone uses the total votes and the percentage cast for each candidate to back out the vote tallies, but only has the percentages to 3 sig figs.
Also explains the truly baroque behavior from 03:04 and 21:59 UTC on the 6th: slowly adding votes to the tally, but the percentage Trump remains at 50.0% (well, between 49.5% and 50.5%…)
What did you study in college?
Sorry, but I have no idea what you’re saying there, so you still haven’t communicated clearly.
You have been misinformed. Haeckel’s embryos are not seriously wrong. The moth experiment is not falsified. And evolution does form a tree (mostly).
As for the rest of it, it’s hardly surprising that a crackpot on one subject would also be a crackpot on another. And you still haven’t addressed much of anything.