
Dr. Winston Ewert put forward his module hypothesis, but I put forward an alternate module hypothesis at the domain and motif level of proteins. It is based actually on papers by evolutionists who have pointed out that the problem of “Promiscuous Domains” remains an unsolved problem in evolutionary biology.
When I put Promiscuous Domains on the table in the Common Design vs. Common Descent thread, the TSZ Darwinists ignored the problem and then declared victory. I viewed their non-response as evidence they didn’t understand the problem and/or preferred to ignore it.
Perhaps pictures are worth ten thousand words. From the NIH, that great source inspiration for the Intelligent Design community, we have the CDART database viewer.
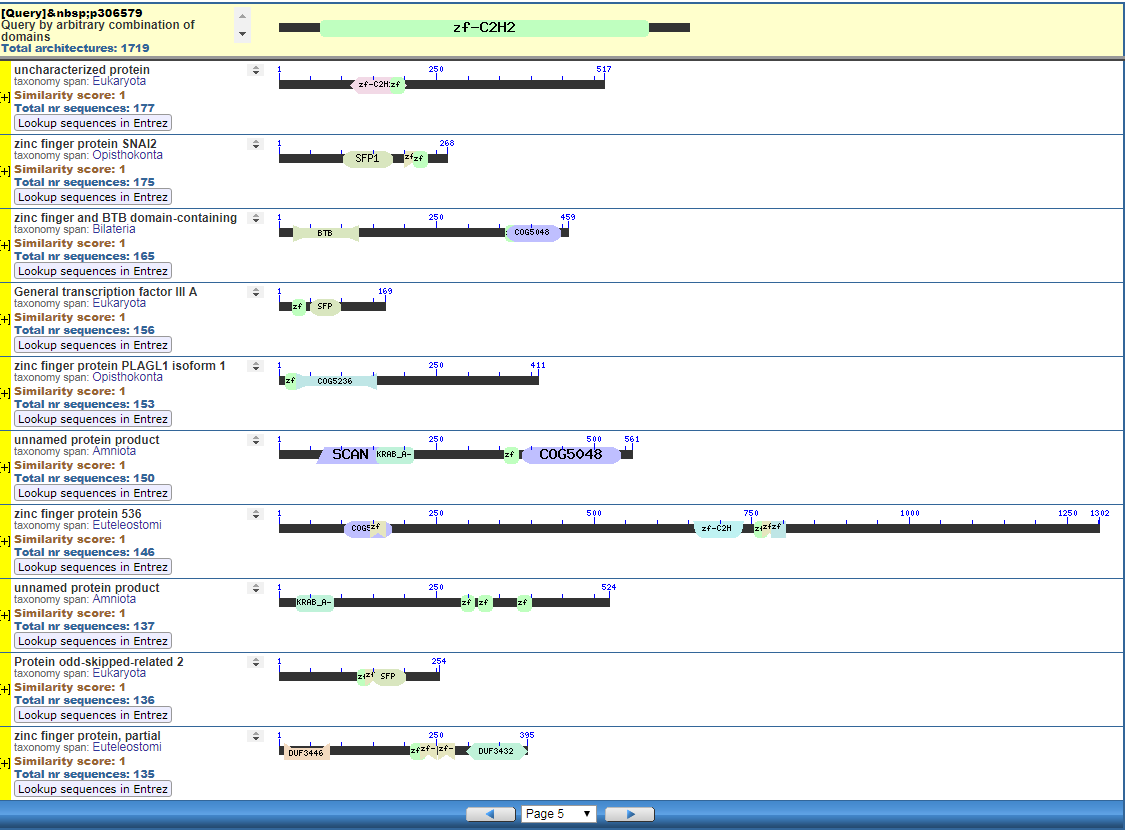
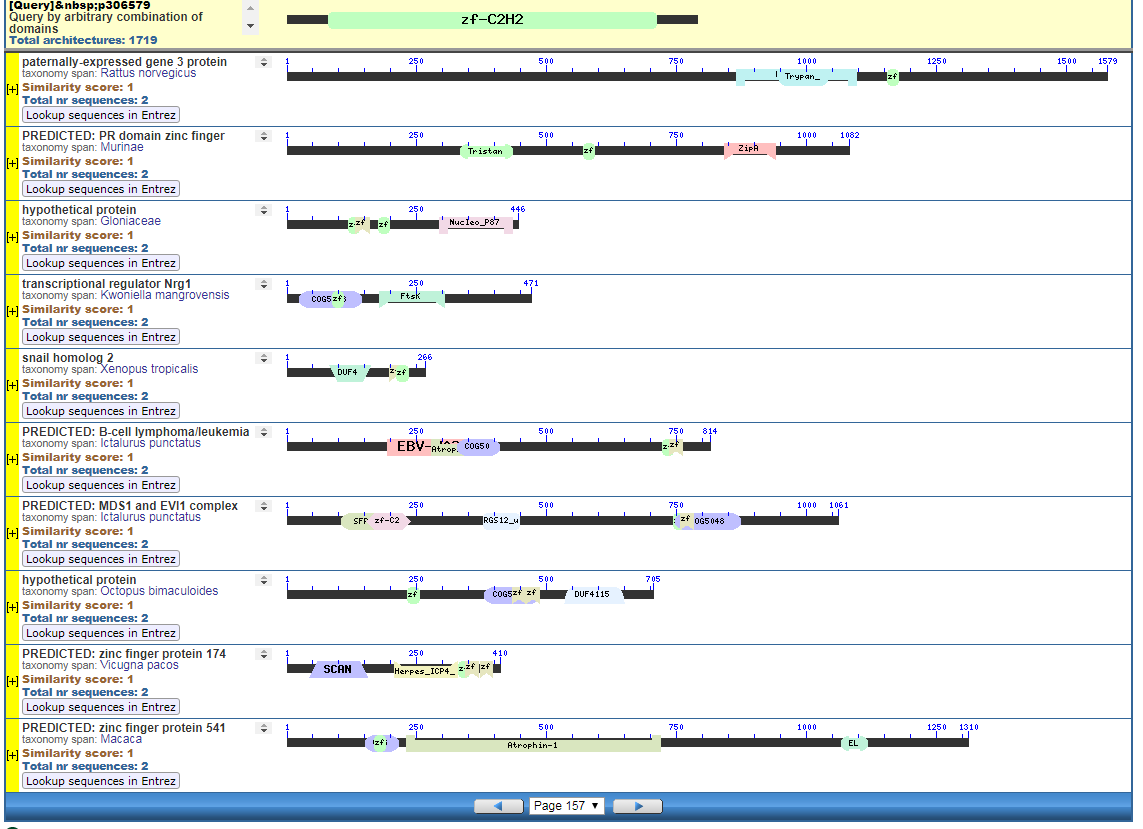
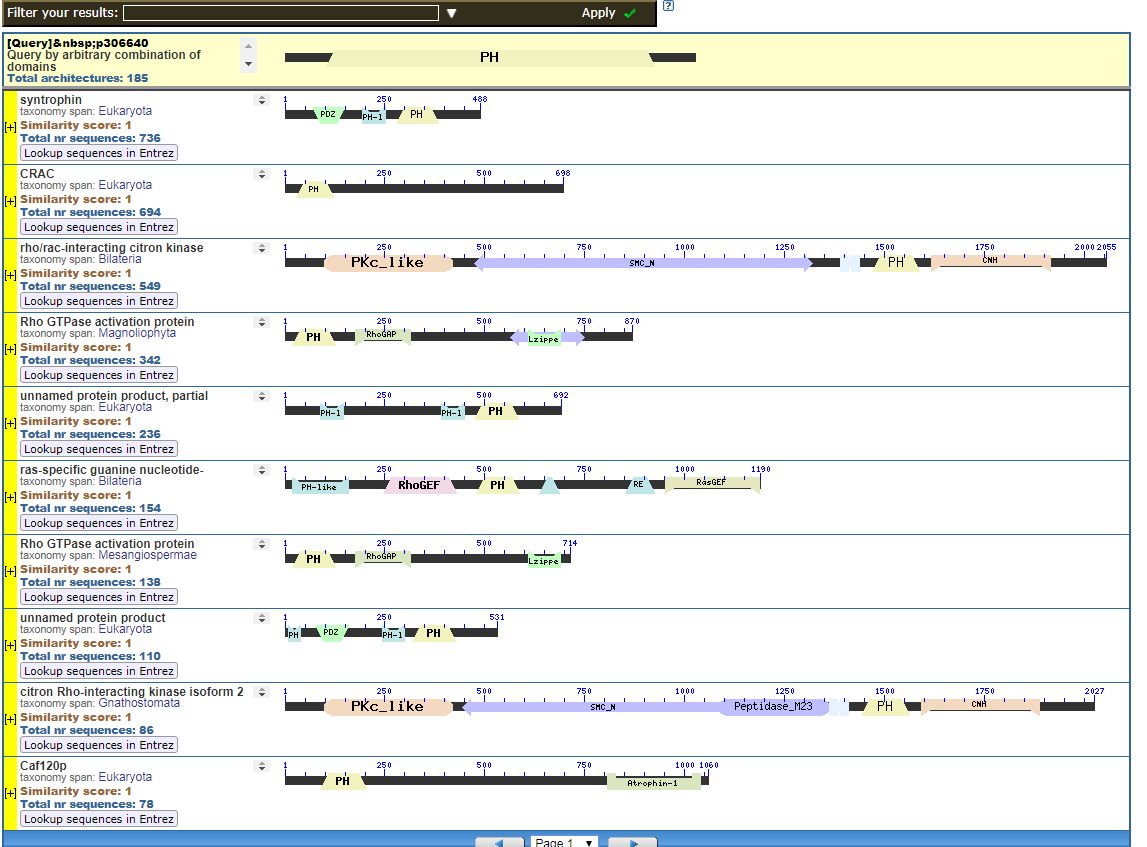
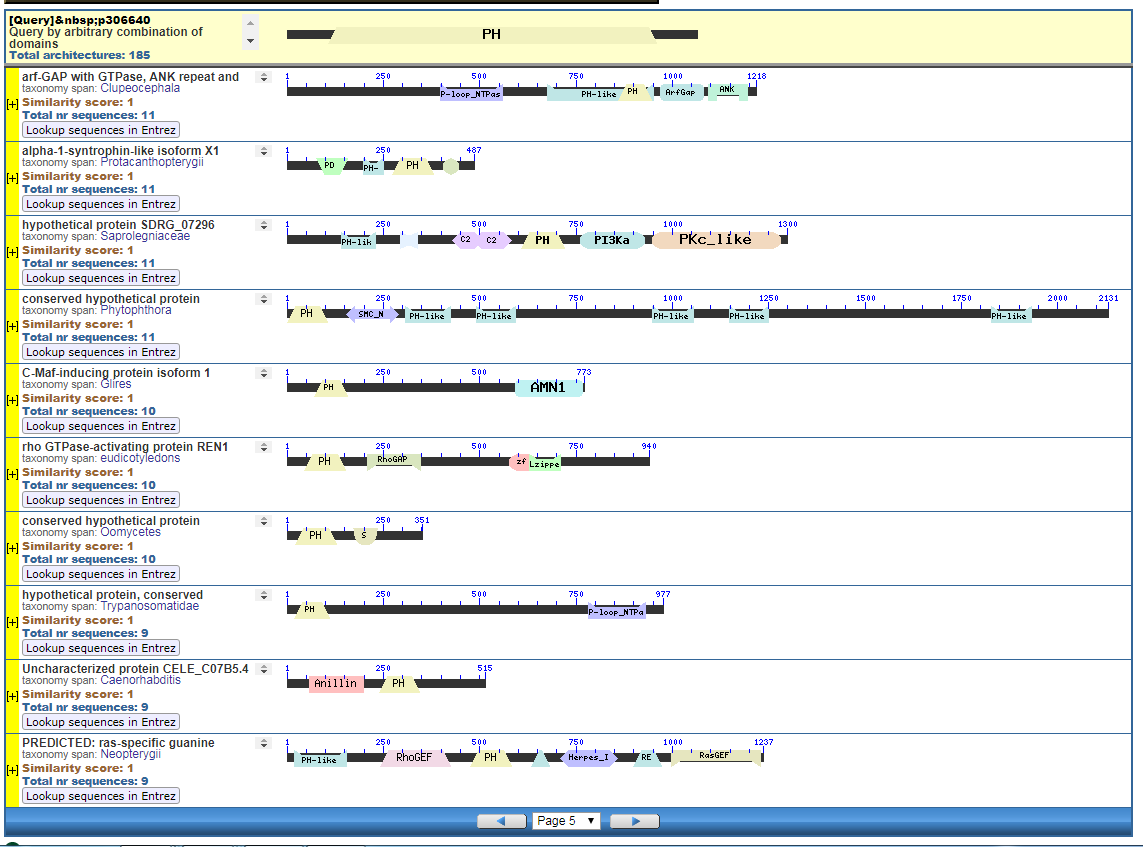
From the CDART viewer, I provide a few of the thousands of diagrams that show the promiscuity of protein domains. The diagrams below show the classical zinc finger ZF-C2H2 “ZF” domain and the Plextrin Homology “PH” domains. Note how the location of domains is “shuffled” to different locations in different proteins. It’s as if proteins are made by different lego-like parts in different order and position. My preliminary look into small 4-amino acid motifs that are the target of phosphorylating kinases suggests the the problem of promiscuity goes all the way down to small motif levels.
Such promiscuity is more consistent with common design than common descent.
Click to Enlarge Classical ZF-C2H2 Zinc Finger Page 5
Click to Enlarge Classical ZF-C2H2 Zinc Finger Page 157
Click to see all CDART Classical ZF-C2H2 Zinc Finger Architectures
This is a very poor start Sal. You just assumed that if someone declares something to be an “unsolved problem in evolutionary biology,” that means that such thing is a problem for evolutionary biology. There’s a difference.
The “problem” of domains is not a problem for evolutionary biology, but a problem in evolutionary biology, and for evolutionary biologists to study. Reason being that evolution, being a natural phenomenon, could not care less about our natural inclination to think in human terms, with simplifications brought from the world of human affairs. Thus, we conceptualize of pieces of proteins as if they were actual modules, and call them “domains.” However, we find difficulty even in trying to delimit the boundaries of said domains, precisely because for naturally recombining pieces of DNA, there’s no real limits. It’s all wherever and however it lands, with natural selection, negative and positive, moving what remains-to-be-seen one way or another. So, fragments covering from unrecognizable to barely recognizable to clearly recognizable, to “complete” domains occur just about anywhere. Your very graphs present that phenomenon. Rather than re-use of designed parts, it looks like fragments of different sizes landing everywhere and anywhere.
Anyway, I’d advice to start by explaining in which way you think that domains are a problem for evolutionary biology, rather than a problem in evolutionary biology. Quoting some claim in some abstract, without understanding what it means (I suspect it was mere hyperbole), is not enough.
Just a suggestion though. I know you IDiots thrive in misinformation and misunderstanding. So, I’d understand why you’d refuse to clarify.
The problem for evolutionary biology is explaining the appearance of domains in different proteins in different locations.
If the appearance of a classical Zinc Finger (there are many families of zinc finger), the domain I chose was the classical zf-C2H2 zinc finger is the result of convergence, then that isn’t common descent is it, it just happens to be a lucky coincidence.
If the appearance of the Zinc Finger or Plextrin Homology domain, or any number of domains is by exon shuffling are any sort of horizontal duplication, one has to explain the coordinated nature of creating functional and integrated proteins that have these domains. Zinc Finger’s a common in chromatin interacting proteins.
In any case, the architecture of these proteins doesn’t follow from any incremental mutational change in a phylogenetic tree does it?
The fact some here don’t understand the problem is evidence they are un-informed of some of the latest problems for evolutionary biology.
I don’t see why would this be a problem. I already explained it in terms of evolutionary biology. Did you read what I wrote?
Here, though very hard to understand, you seem to be a tad mistaken. If you put things in false dichotomy terms (common design vs common descent), and then use definitions at whatever level is convenient to your beliefs, then you’re not going to go very far in understanding.
If the zink finger of your predilection is the result of convergent evolution, that means that they’re still the result of evolutionary phenomena, not of common “design.” See how that’s not a simple dichotomy?
If the fragment comes from exon shuffling, then the apparent “coordination” is nothing but the natural result of the thing exchanged being an exon. Exons are bounded by very large introns, and thus the probability of recombination within such large piece of non-coding DNA is huge. That would give the appearance of a real boundary to the domain, but would still be explainable as natural phenomena.
If the domains recombine in other ways, then the “coordination” is but your interpretation. Still much better explained and much more compatible with natural phenomena, rather than with “design.” Again, check those “architectures.” Don’t you see the differences in length among those supposed domains? That’s what we expect to get from unplanned recombinations, with some cleaning up by natural selection, both negative and positive.
So what? It still follows from naturally, verified to occur, phenomena, such as recombination, plus negative and positive selection. Also note that those zink fingers are not identical in sequence, which means that they also suffer from “incremental mutational change.”
This is an interesting claim, since it seems like it’s you who’s looking at this from a very shallow perspective. Check the coordinates of those domain matches. Check their differences in length (it’s noticeable in the graphs already). Check how much they cover of the “official” domains they’re supposed to match. You’ll find that they don’t look that “modular” after a careful look.
I’d advice to read more carefully the comments you’re supposed to be giving answers to. I repeated quite a bit from my previous comment because you seemed to miss those explanations.
Entropy thinks he’s explained things. That’s a howler.
How is this a consequence of common design? It would seem to be the same “re-use” of parts that Bill keeps going on about. But if these are re-used parts, why aren’t their sequences identical among instantiations? Why do these zinc-finger protein/gene sequences fit a nested hierarchy? Why do different loci tend to fit the same nested hierarchy? It doesn’t add up.
As I said, you, like all IDiots, thrive on misunderstanding and misconceptions, which is why I wasn’t expecting much from you, let alone that you’d read my explanations.
Ciao Sal.
To optimize scientific discovery. Did you ever look at alignments of zf-C2H2 alignments. It’s pretty obvious what the critical residues would be in a 3D conformation. That would not be evident if the zf-C2H2 fingers were identical.
It would seem the designer had us and our capabilities to comprehend in mind.
stcordova,
Hi Sal
I don’t understand the point you are making.
I referred to motifs. Some of the motifs are surprisingly small, as in 4-amino acid sequences. That’s just long enough to substantially reduce the probability of an errant protein binding interaction by a kinase due to a random sequence.
Observe this segment from a pair of TopIsomerase2A and TopIsomerase 2B isozymes each coded on SEPARATE human chromosomes.
Click to Enlarge
The “rkPs” motif in Top2A does not appear in Top2B, and is targeted by the kinase CSN2KA1. In contrast the “rkAs” motif appears in Top2B and not in Top2A, but as of yet I’ve not been able to determine the kinase responsible for phosphorylating the Serine amino acid.
Ok, now go to this PhosphositePlus website here and enter “rkPs” and you see the motif “promiscuously” spread across proteins apparently as phosphorylation targets of kinases that recognize the “rkPs” motif.
https://www.phosphosite.org/psrSearchAction
You can see that only about 500 proteins spread across humans, rats, mice, cows are known kinase targets. Kinases are “writers”. Phosphotases are “erasers” and other binding partners are “readers”. Are those kinase targets accidents or are they real. If they are real, and there is reason to believe there are numerous such post-translational targets in the genome, then that’s a problem for evolution by random mutation.
Bill Cole might appreciate these targets as they are somewhat like specialized ports (USB, microUSB, ethernet, PCI, whatever…).
The “rkPs” motif and the “rkAs” motifs don’t appear in yeast, fruitflies, nematods, Arabidopsis Thaliana. So how did those motifs evovle? We can put the TopIsomerases in a nested hierarchy, right, but what if they are packed with species specific regulatory motifs. That doesn’t exactly suggest a random walk where most of the amino acid are of no consequence. Quite the opposite.
Start with the diagrams. Each row in each diagram represents a protein, usually one that is shared among species. Note the different colored boxes in the rows, they are like legos. Note how each protein has a different arrangement of legos.
In the first two diagrams, note how the light-green “zf” box is position in different places in various proteins. The “zf” is a reusable conceptual component, somewhat like the idea of “tire” or “spark plug” in cars. Much like there are different tires, there are also different “zf” parts, but the zf construction is recongizable as a separate functional/conceptual entity.
i can’t keep up with this as it demands a schiolarly attention that includes basic knowledge in these matters.
YET common design easily can beat common descent using the same data.
there never is a need to imagine common descent because the historic common design idea is better.
we all have eye balls because god created on creation week, in kinds, eye balls based on a eye vall blueprint. a great idea very much mimicking physics in principles.
evolutionism denies biology is like physics because they too quick ignored common laws as a guiding light.
Anyways a good contribution to the intellectual investigation.
That’s an interesting hypothesis. I wonder how you would test it.
But it doesn’t explain why this variation is structured as a nested hierarchy. For your purposes, randomly structured variation would do as well.
[Citation needed]
So can you link some of these papers that describe the nature of the problem?
stcordova,
Hi Sal
Among your 4 AA motifs the main differences appear to be L versus D configurations. Do you know how the cell converts from L to D?
@Sal
So going back to the common descent thread we find you making this statement regarding the “Promiscous domains problem”:
And I’m thinking this sounds like a deliberately silly strawman? Who actually thinks this willy-nilly stuff(citations please), and how does that describe the way in which these domains is thought to have evolved?
It seems to me the great majority of random insertions would be nonfunctional (or at least deleterious), but occasionally one might work and be beneficial. How often? Well who really knows, but why should we believe it would be rare enough to be a problem for evolution? Do you have some sort of special knowledge that allows you to conclude that the number and distribution of them seen in life, is a problem for evolution? I hope this is based on more than just some hunch or intuition you have.
How would the the motifs being non-identical “optimize scientific discovery”? Do you have access to multiple alternative biospheres where other sentient beings used science to investigate biological phenomena and through that you have collected data that allows you to conclude that our particular biosphere and it’s biochemistry is optimized towards scientific discovery?
Please explain how a motif not being identical between different loci is optimal or superior for scientific investigation? And then please show some compelling empirical evidence that supports that conjecture. You wouldn’t just be making shit up because it sounds pleasing as a creationistic rationalization, right?
Especially because the stats fail all too early, thus preventing us from detecting divergent domains by swallowing them in an ocean of false positives. Designed for optimal scientific discovery? Yeah. Right. Seems like natural divergence wins this round hands down as well.
Not that Sal would care.
E.T.A. Good designers would leave some blueprints instead, Wouldn’t they?
One of the first papers on promisuous domains:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2722818/
and
Identified in a sister paper is the PH (plextrin homology) domain. I provided in diagrams 3 and 4 some of the CDART architectures with Plextrin Homology domains:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2259109/
Hi,
The motifs “rkPs” and “rkAs” are not involved in conversion of L to D to my knowledge but are used as memory addresses for the “s”. The memory is “written” to by a protein known as a kinase. Flipping this “s” affects the fold of the protein as well as its interaction partners.
Go to this website
https://www.phosphosite.org/psrSearchAction
and enter, “rkps” as shown here:
When I enter “rkps” at the above link, I get a listing of proteins that have this “rkPs” motif. Here is a sample screen shot of the 593 or so proteins at Phosphosite where rkPs is a target of the CSNKA1 (CK1).
So how did this motif get inserted into proteins. It’s not exactly a good thing for accidental phosphorylations to happen, so it’s not desirable for proteins to randomly adopt motifs that could become undesirable phosphorylation targets, not to mention, if the phosphorylation is “read” by another protein, it’s not good the reader is reading a phosphorylation by accident.
stcordova,
If you look at the chart above and RKPS on the right you will see some with RKPs with the small s in red. Does the small s stand for a D type amino acid?
It means it is confirmed as a phosphorylation target. Almost all amino acids in proteins are L type with very very few exceptions.
But, your question was extremely helpful as I didn’t notice the lowercase until you mentioned it. So I looked it up.
Your question alone and your observation was worth the trouble of posting this at TSZ, so a big thank you for improving my understanding!!!!
The blind misleading the blind.
As if you’ve demonstrated any knowledge in this realm, so before you boast, you might try to show to us you actually know what you’re talking about.
Oh, I seem tor recall you were skeptical of “readers”, “writers,” and “erasers” in biology and were so negative on the “-omes” of which the phosphoproteome is a rather important “-ome”.
stcordova,
Sorry, Sal. The temptation was too strong.
A picture is a worth a thousand words, here is a map of the publications studying the post-translational modifications on Top2A alone. The Y-axis is number of publications so far:
Click to Enlarge
The CLUSTAL and MUSCLE alignments across species shows the “rkps” motif in Top2A seems mostly restricted to placental mammals (in my tiny sample space). One can see it doesn’t exist in chicks or yeast.
Click to Enlarge
One of the interacting partners with Top2A is a protein known as INTU. It also has the “rkps” motif, but has not been confirmed yet to be a phosphorylation target. A testable chemical prediction is that there is a context where Top2A and INTU are co-regulated by phosphorylation of the “rkps” motif.
The MUSCLE and CLUSTAL alignments show that “rkps” in INTU is mostly in placental mammals.
Click to Enlarge
A testable chemical prediction is that when “rkps” is mostly conserved in mammals it will be a phosphorylation target.
The difficulty for evolution by random mutation is if this motifs are conserved in placental mammals in multiple proteins AND are phosphorylation targets. This would imply the motif had to co-evolve simultaneously in multiple proteins when mammals came to be.
What would be difficult for random mutation to explain is the simultaneous and sudden appearance of the same functional motifs in multiple proteins in the same taxonomic group.
You’re just blathering. What are we supposed to conclude from all this and why?
So it’s largely unknown because, as the paper says, there’s not much direct evidence of which of several known mechanisms. And?
Transposition? Exon shuffling? Duplication into another gene? Those are mechanisms that could do it and are known to actually happen.
Generally no, just like it is generally not a good thing for mutation to happen because most mutations are deleterious. But they still do happen, and on occasion one is beneficial. Why should it be different with various motifs and domains? Do you have some special knowledge that allows you to conclude this isn’t possible?
You are stuck in black and white thinking again.
… and?
… sooo, what?
And?
Why is that a problem?
And?
Why does it have to be simultaneous and sudden? Just because you can identify a motif as primarily existing in some proteins in mammals doesn’t mean it had have popped up all at once at the same time. It just doesn’t follow.
That’s but a sentence in the introduction, yet the authors wrote a section, three paragraphs long, explaining exactly that. The paper is a review, so I really don’t know why they talk about that as a mystery in the abstract and intro, other than for over-selling their review. I told Sal that he has to read the articles themselves beyond the abstract. He searches for words and quotes regardless of anything else.
The second paper quoted by Sal is about the evolution of those domains. Yes, with explanations. Both papers from 2008/2009, citing articles from 2001 that contain models of the very domain evolution that Sal thinks is a problem for evolution. So, what mystery is that? Oh. Of course! The mystery is how can Sal be in a PhD program and still fail to read articles for comprehension? It’s customary of Sal to quote from articles that contradicts his claims. His illiteracy shall become legendary. An exemplar of what it takes to belong in the IDiot movement.
This is a good introduction to phosphorylation of proteins:
https://en.wikipedia.org/wiki/Protein_phosphorylation
I’m trying to show the density of functions of the protein. That’s about 100 post translational sites in protein that’s about 1600 residues long. You think that’s mostly noise or serves some function, like say signalling and positioning? I forgot to mention, somewhere toward the right side C-terminus is a Nuclear Localization Signal, so even what seems like a innocuous amino acid sequence whose function in IN VITRO isolation might not have significance has a lot of IN VIVO significance in navigating the protein to the right location in the cell. FWIW:
https://en.wikipedia.org/wiki/Nuclear_localization_sequence
So, when the ancestor of eukaryotes which had no nucleus came to be, how did proteins go in and out of the nucleus without Nuclear Localization Sequences Signals (NLS)? I mean, wouldn’t you think they would, eh, sort of die?
If the NLS is different between major species, wouldn’t you think that’s a problem. What is the mechanism of nuclear localization.
The question then could be extended to all sorts of positioning and navigation questions as to the possible role of post-translational modifications in positioning proteins.
Yeast Top2 is only about 40% similar to Human Top2A. We can of course be more formal about this, but a guestimate would be 60% of those post translational modifications changed between yeast and human. Are you ok with such a radical change in cell signaling architecture for just one measily protein?
The problem is fusing those domains in a coherent way that functionally works with the rest of a system. By way of analogy, one doesn’t make a car part randomly, it has to fit into the rest of the car geometrically and functionally to make the part coherent.
Here is a picture of the SET domain and Zinc Finger domains in various proteins that are Chromatin/Histone modifiers. These proteins “write” on the memory locations on the histones. On the left is the name of the protein (“writers”), on the right is the specific memory location that is modified. These are post-translational modifications, by the way.
You think such integration of these proteins can happen by random accidents? I don’t think that is the natural expectation:
Sal,
Perhaps a better taxon sample would illuminate a few things. You talk about placental mammals yet you have no marsupials or monotremes, and a poor sample even of placentals. If you want to investigate the evolution of a locus (and this is indeed evolution), then you need to sample more densely. Unfortunately, there’s a couple hundred million years worth of unbroken branch between the ancestral node of living synapsids and the ancestral node of amniotes, so good luck for anything in that time range. Still, I note that Xenopus has KKPS at the sites you’re on about, in both genes. That might be a hint that the site, in some form, has been around with a similar function for longer than you think.
What is the color coding in your alignments? I can’t make any sense of it.
What function does “common” have in your title except to mimic the term “common descent”? What does the term “common design” actually mean? You need to flesh out your model before it’s possible to test it. If you refer to separate creation of species, that’s one thing. Separate creation of some nebulous higher taxonomic grouping is another. And how do you rule out common descent with occasional divine intervention to confer particular mutations?
So far you’re just putting up a bunch of colorful pictures and shouting “wow”. Make some kind of argument for some particular model in contrast to some other particular model.
Great suggestion!
The color coding is MEGA’s, nothing that I did.
Ok, I think I’ve established there are domains that are promiscuous. That is my conclusion and that of evolutionary biologists. So life at the protein level is modular. Is the modularity the result of common descent or common design. I don’t think randomly shufflling modules is a way to make coherently integrated proteins, especially those that target specific residues on specific histones such as in the diagram above (the SET and zinc-finger domain containing proteins).
So then back to John Harshman’s question about “why the nested hierarchical pattern.” At ICC 2018, I talked with my colleagues and insisted if life is young, then the hierarchical pattern is by design. The question is why? My answer: God wanted to make it easier to decipher biological function.
I argued, if we did not have the hierarchical pattern, especially in proteins sequences across species, but rather if the Designer created the same protein the same way in every species, we wouldn’t be able to identify critical features. What are some critical features:
1. catalytic sites (conserved across species)
2. metal binding sites (conserved across species)
These critical sites help identify domains (the Zinc Finger zf-C2H2) is a real easy one. So thus domains can be identified because of the patterns of diversity and similarity. If there were no hierarchical pattern we could not identify them so easily.
So, what about features conserved in groups but not across the entire spectrum of species with that protein? By way of extension, this suggests somewhat lesser but important features like targets of post translation modification. Some have suggested that this could be used to identify 3D structural features, and I’m part of a project that is exploring this. My current interest is using the patterns of similarity to identify post translational targets or localization signals (like nuclear localization).
That said, here is someone I met at ICC 2018 who studied evolutionary biology. He relates very well why I think the fossil record is young and the result of a Cataclysm, not millions of years of slow sedimentary accumulation. He explains it in the first 4 minutes or so:
No, that doesn’t work, as I have already explained to you. If God wanted to make it easy to decipher biological function (by highlighting only crucial bits as invariant, apparently), all he would need would be randomly patterned — which is to say unpatterned — variation among species. A nested hierarchy pattern is unnecessary, not to mention the same nested hierarchy at different loci.
Even if you somehow manage to support the notion that God is necessary to explain the existence of some innovations, that doesn’t distinguish separate creation from common descent with guided evolution. And it certainly doesn’t support recent creation, which is out of the question given the evidence from every field of science.
It’s more subtle than that, what if the Designer wanted to highlight multiple functions, he could do so with a hierarchical arrangement. Random variation will not work, btw, for co-regulated motifs spread across multiple proteins, as I was trying to show with the PhosphoSite motif search.
Many of the phylogeny programs don’t deal with the creation of “promiscuous” domains either with models of little point mutation and small indel changes, so promiscuous domains and motifs don’t fit most traditional single protein or DNA segment phylogenetic analyses, especially domains and motifs spread across multiple proteins simultaneously in a way that facilitates coherent co-regulation.
The evidence of promiscuous functional domains and motifs argues against random mutation. Random variation carries with it expectation values and outcomes, and coordination isn’t an expected outcome. That’s why tornadoes passing through a junkyard don’t create functioning 747 jetliners.
In any case, thanks for participating in the discussion. Your objection for the weak taxonomic sample was an excellent one, and so was your complaint about the color scheme of MEGA.
Here is an alignment of the most diverse zf-C2H2 zinc fingers in terms of sequence from the NIH CDD. The critical Cysteine “C” and Histidine “H” are clearly obvious. These are the residues that are connected to the Zinc Ion, making the protein a “metal protein”.
Click Here To ENLARGE
Here is a 2D and 3D rendering of the Zinc Finger showing the positioning of the critical residues in the alignment in the previous comment:
Click to ENLARGE
Probably for another thread, I only watched the first 4 minutes, as you recommended. This seems to be his logic: because we found a fossil that looks like it was buried rapidly, all fossils are buried rapidly and not over millions of years. A classic case of building a strawman and knocking it down.
The time it takes to fossilize an organism is only very loosely related to the time it takes to lay down the totality of the strata that we find fossils in. It is perfectly possible for a fossil to be created in a comparatively short time, to be embedded in rock layers that in their totality have taken very much longer to form. As to the argument that you don’t find neat dead fish at the bottom of the ocean – if you take a sediment core down from the ocean floor you will find dead organisms and fossils right from the top, and all the way down to the end of your core (unless you drill through some igneous or metamorphic rocks). In fact, some sediments consist almost entirely of dead organisms.
KRAB domains are restricted to tetrapods.
Several proteins have KRAB domains. Note how there are MULTIPLE The Zinc Fingers on the protein. Their structure is designed to target specific DNA motifs as these proteins are often transcription factors and have to park on the right spot on the genome.
Are KRABs promiscuous? Or is one going to argue all these transcription factors with KRABs in them descended from a common ancestor? Well if one argues that, what about the genes these KRAB-containing transcription factors are associated with? This would seem to require a lot of coordinated co-evolution to get the KRABs all over the proteome.
Anyway this picture was too good to pass up:
Here was an old article that regrettably has very little similar follow on research. As one can see from the previous comment, some proteins have MULTIPLE zinc fingers. Alternative splicing can modulate the Zinc Fingers present. Note that each zinc finger is a little different from another, and hence has slightly different function.
Alternative splicing can direct which zinc fingers combinations appear, and this has developmental consequence. Ergo, I don’t think unguided evolution can randomly shuffle domains and get something coherent.
A reasonable explanation for the spread of zinc fingers of many varieties over many proteins implementing coherent function is common design, not common descent:
https://www.ncbi.nlm.nih.gov/pubmed/1411512
A lot can happen in four billion years.